
反胶束萃取对大豆分离蛋白结构和特性的影响
2019-05-05 06:56陈复生
食品科学 2019年7期
张 倩,陈复生*
(河南工业大学粮油食品学院,河南 郑州 450001)
大豆是一种富含油脂和蛋白的重要经济作物,是世界上种植最为普遍的作物之一。大豆中约含有20%的油脂、40%的蛋白质和30%的碳水化合物,常作为一种重要的植物油料作物用于食用油的生产,副产物的主要成分是大豆蛋白。大豆蛋白氨基酸种类齐全,且人体必需氨基酸比例较高,不含胆固醇,具有较高的营养价值。此外,由于大豆蛋白来源广、成本较低且具有较好的加工特性,因此被广泛应用于食品行业[1-2]。目前大豆蛋白的提取方法主要是碱溶酸沉法,该方法在蛋白提取过程中会产生大量酸、碱废水,蛋白活性也会受到影响,但因其适用范围广、工艺相对较为成熟,依然是主流的蛋白提取方法。
反胶束作为一种新型的高效分离技术也逐渐被大众所熟识,近年来关于反胶束体系的研究有很多[3-6]。反胶束是表面活性剂在有机溶剂中自发形成的胶束结构,疏水性尾部朝外、亲水性头部朝内,形成类生命环境“水核”结构[7-8]。该体系能够同时高效地分离油脂和蛋白,且能最大程度上保证蛋白质的生物活性[9]。因此,反胶束体系比较适用于油料作物油脂和蛋白的分离,在工艺流程上能有很大的简化。
近年来,有很多关于反胶束萃取和纯化蛋白质的研究,以及该方法与传统碱溶酸沉法所提取得到的蛋白质在结构和其他理化性质方面的差异研究[10-12]。本研究系统地对比了两种方法(反胶束法和碱溶酸沉法)分别提取的大豆分离蛋白样品在结构和热力学、流变学特性上的差异,分析反胶束提取对大豆分离蛋白结构和特性的影响。
1 材料与方法
1.1 材料与试剂
全脂大豆粉 河南省鲲华生物技术有限公司;丁二酸二异辛酯磺酸钠(sodium bis(2-ethylhexyl)sulfo succinate,AOT) 上海海曲化工有限公司;异辛烷(分析纯) 天津市科密欧化学试剂有限公司;Na2HPO4、KH2PO4、KCl、浓硫酸(分析纯) 洛阳昊华化学试剂有限公司。
1.2 仪器与设备
BS210S型分析天平 德国Sartorius公司;GL-20C型高速冷冻离心机 上海安亭科学仪器有限公司;Zetasizer Nano ZS90激光粒度分析仪 英国马尔文仪器公司;HAAKE MARS 60动态流变仪、WQF-510A傅里叶变换红外光谱仪 美国赛默飞世尔科技公司;Cary Eclipse荧光分光光度计 美国Varian公司;Q20差示扫描量热仪 美国TA Instruments公司;DYY-6D电泳槽北京市六一仪器厂。
1.3 方法
1.3.1 反胶束法制备大豆分离蛋白
本研究采用的是AOT/异辛烷反胶束体系。提取方法参考文献[13]。称取一定质量的AOT于锥形瓶中,加入一定体积的异辛烷配制成0.06 g/mL的溶液,摇匀至AOT完全溶解,然后加入含0.05 g/mL KCl的磷酸盐缓冲液(用2 mol/L盐酸溶液调节pH值至8.5),气浴振荡1 h后静置12 h,若溶液澄清透明即为反胶束溶液,若溶液不澄清,则重新配制。取一定体积的澄清反胶束溶液,按照质量浓度0.02 g/L加入一定质量的豆粉,40 ℃水浴振荡30 min,之后4 000 r/min离心20 min,取上清液,加入等体积的含1 mol/L KCl的磷酸盐缓冲液(pH 8.5),离心后下层溶液即为溶有大豆分离蛋白的溶液。对该溶液透析冻干后即得到反胶束法制备的大豆分离蛋白。
1.3.2 碱溶酸沉法制备大豆分离蛋白
参考Zhao Xiaoyan等[14]的方法,首先在分离蛋白前需要对全脂豆粉用正己烷(质量比1∶10)进行脱脂处理。取一定量的脱脂豆粉与0.05 mol/L磷酸盐缓冲液(pH 8.5)按照料液比1∶20混合,40 ℃水浴振荡1.5 h后4 500 r/min离心20 min,将上清液留作后用,沉淀重复上述操作。将两次得到的上清液混合,用2 mol/L HCl溶液调节pH值至4.5后4 500 r/min离心20 min,弃去上清液,将沉淀复溶后透析冻干,即得到碱溶酸沉法制备的大豆分离蛋白。
1.3.3 十二烷基硫酸钠-聚丙烯酰胺凝胶电泳分析
将蛋白粉末溶于去离子水中配制成质量浓度2 mg/mL的蛋白溶液。十二烷基硫酸钠-聚丙烯酰胺凝胶电泳(sodium dodecyl sulfate-polyacrylamide gel electrophoresis,SDS-PAGE)操作参照文献[13]。
1.3.4 傅里叶变换红外光谱测定蛋白的二级结构
通过KBr压片法测定蛋白样品的透射率的变化趋势。每个样品重复3 次。重点将对酰胺I带(1 600~1 700 cm-1)范围内的谱图通过PeakFit软件处理。
1.3.5 表面疏水性的测定
表面疏水性的测定采用8-苯胺基-1-萘磺酸荧光探针法,参照文献[15]进行操作。
1.3.6 二硫键含量的测定
二硫键含量的测定采用Ellman’s比色法[16],总巯基含量与自由巯基含量的差值为二硫键含量。自由巯基含量的测定:取0.5 mL 10 mg/mL蛋白溶液加入2.5 mL Tris-Gly缓冲液(0.086 mol/L、pH 8.0),加入0.02 mL Ellman试剂(含4 mg/mL 5,5’-二硫基双-2-硝基苯甲酸的Tris-Gly溶液),振荡后在25 ℃保温60 min,然后在412 nm波长处测定蛋白溶液的吸光度。总巯基含量的测定:在0.2 mL蛋白溶液中加入0.02 mL β-巯基乙醇和1 mL 10 mol/L尿素溶液(溶于Tris-Gly缓冲液),在25 ℃保温60 min后再添加10 mL质量分数12%三氯乙酸,25 ℃保温60 min后离心。用5 mL三氯甲烷溶液清洗沉淀物两次,将沉淀物溶于8 mol/mL尿素中,再加入0.05 mL Ellman试剂。测定时以未添加蛋白质的溶液作为空白对照,在412 nm波长处测定吸光度。
1.3.7 蛋白粒径的测定
根据Guo Juchen等[17]的方法,采用Zetasizer Nano ZS90激光粒度分析仪对反胶束体系的粒径和蛋白体系的多分散系数进行测定。
1.3.8 热力学特性的测定
参照文献[18],采用Q20差示热量扫描仪对蛋白质的热变性以及玻璃态转变温度进行测定。热变性的测定:在铝盒中称取3 mg蛋白样品,用移液枪移取10 µL 0.05 mol/L磷酸盐缓冲液(pH 7.0),加盖密封后室温静置过夜。测定前首先在25 ℃下稳定5 min,然后从25 ℃升温到110 ℃,升温速率为5 ℃/min。玻璃态转变温度的测定:在铝盒中称取一定质量的蛋白质粉末,从-10 ℃开始升温,升温速率为10 ℃/min,首次升温到略高于玻璃态转变温度,降温后再次加热至180 ℃。
1.3.9 凝胶特性的测定
采用动态流变仪对蛋白溶液的凝胶特性进行测定。将蛋白样品溶解于0.5 mol/L磷酸盐缓冲液(pH 7.6)中,配制成质量分数11%蛋白溶液。室温下搅拌3 h后4 ℃静置24~36 h,使蛋白充分水化,测定前室温搅拌平衡30 min。采用椎板测定,间隙为0.05 mm,温度变化过程为:以5 ℃/min从25 ℃升温至95 ℃,在95 ℃保温30 min,以5 ℃/min降温至25 ℃。观察在温度变化的过程中储能模量(G′)和损耗模量(G”)的变化规律。对所成凝胶进行频率扫描(频率为0.1~10 Hz,应力为1%),进一步对蛋白凝胶的状态进行表征。
1.4 数据统计与分析
实验数据采用SPSS 16.0软件进行单因素方差分析,P<0.05表示差异显著。采用Origin 8软件作图。
2 结果与分析
2.1 SDS-PAGE结果分析
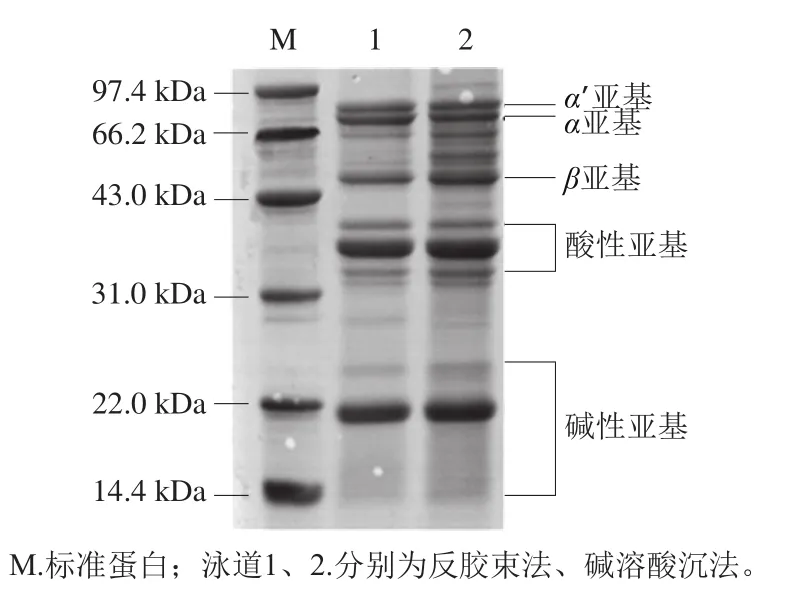
图1 两种方法分离提取得到的大豆分离蛋白的SDS-PAGE图Fig. 1 Sodium dodecyl sulfate-polyacrylamide gel electrophoresis profile of different SPIs
如图1所示,两种方法分离的大豆蛋白的条带对应的分子质量和标准蛋白吻合,因此可以确定两种方法得到的蛋白样品是大豆分离蛋白。对比两种方法可以看出,碱溶酸沉法得到的大豆分离蛋白的α亚基有部分的解离现象,条带减弱,分解成分子质量较小的条带。这说明反胶束法相较于传统方法而言更能保持蛋白分子的生物活性。
2.2 傅里叶变换红外光谱定量分析蛋白的二级结构

图2 两种方法得到的大豆分离蛋白的傅里叶变换红外光谱图Fig. 2 Fourier transform infrared spectra of different SPIs
如图2所示,酰胺I带的波数范围为1 600~1 700 cm-1,是由肽链上的羰基(C=O)结构中双键的伸缩振动引起的,该振动能在一定程度上表征蛋白质的结构特征[19-20]。因此对蛋白质二级结构的表征重点分析该酰胺I带的谱图[21]。反胶束法和碱溶酸沉法分离得到的样品的酰胺I带对应的波数分别为(1 653.0±2.9)cm-1和(1 653.0±1.1)cm-1,没有明显差异。通过对谱图进一步去卷积求导拟合,得到具体二级结构的含量见表1。

表1 两种方法得到的大豆分离蛋白的二级结构含量Table 1 Contents of secondary structures in different SPIs
蛋白质二级结构的定量分析可以通过求二阶求导的方法实现,酰胺I带范围内的二阶导数拟合曲线与蛋白质二级结构的含量呈线性相关[22]。从表1可以看出,反胶束法得到的大豆分离蛋白的β-折叠含量明显低于碱溶酸沉法,但β-转角结构含量有所增加,这很有可能是两种结构之间发生相互转化。β-折叠相较于α-螺旋结构而言是一种舒展的结构,是通过众多链间氢键相连接的稳定的片层结构,而β-转角则是肽链伸展方向产生180°转变处的分子结构[23]。因此,反胶束法得到的大豆蛋白β-折叠含量的降低和β-转角结构含量的增加将直接导致蛋白分子稳定有序链层结构减少,整个分子的紧实性降低[24],分子所占空间增加。而碱溶酸沉法得到的大豆蛋白的β-折叠含量较高,说明蛋白分子的展开程度更高。二级结构的差异一方面表明不同的提取方法会对蛋白质的结构产生影响;另一方面,结构的差异也会影响蛋白产品的相关性质,如溶解性、热稳定性、表面疏水性、成胶性等。
2.3 蛋白质表面疏水性和二硫键含量的变化
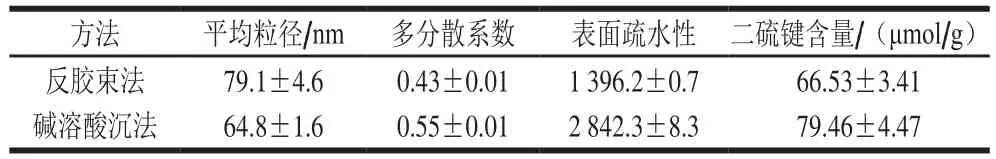
表2 两种方法得到的大豆分离蛋白的平均粒径、多分散系数、表面疏水性和二硫键含量Table 2 Average particle size, polydispersity index, surface hydrophobicity and disulfide bond content in different SPIs
如表2所示,碱溶酸沉法得到的大豆分离蛋白对应的表面疏水性明显高于反胶束法。表面疏水性的变化是蛋白质在提取过程中分子结构展开的程度不同导致的。碱溶酸沉法得到的大豆分离蛋白的分子结构展开更明显,更多的内部疏水基团暴露出来;而反胶束方法能够最大程度上保持蛋白质的分子链折叠结构,这也再次从结构上证实反胶束体系独特的“水核结构”能够保持蛋白质的生物活性。此外,出现这一差异的原因很可能是由于二级结构的差异,传统方法得到的大豆分离蛋白含有较多的β-折叠结构,而β-折叠结构具有较高的疏水特性,因此,表面疏水性会随着β-折叠含量的增加而升高[25-26]。这些结构上的差异将直接影响蛋白质的凝胶性、乳化性、溶解性等。
如表2所示,反胶束法分离得到的大豆蛋白的二硫键含量低于碱溶酸沉法,和Zhao Xiaoyan等[13]的研究结果是一致的。宏观上看是由于不同方法得到的大豆分离蛋白的7S和11S亚基的含量不同,导致蛋白质分子中的二硫键含量也存在差异;从分子结构上看主要是由于反胶束环境会对蛋白质表面的巯基和二硫键产生作用,破坏二硫键的形成[27-28]。
2.4 蛋白质分子粒径的变化

图3 两种方法得到的大豆分离蛋白的粒径分布Fig. 3 Particle size distribution of different SPIs
从表2中平均粒径的结果可以看出,反胶束法得到的大豆分离蛋白的平均粒径要大于传统碱溶酸沉法,这和Boulet等[29]的结果一致。且两种蛋白的粒径分布范围都比较宽,多分散系数都在0.5左右,因此,参考平均粒径的结果,重点通过粒径分布图来分析两者粒径之间的差异。图3显示了基于体积分数的粒径分布,可以明显看出,反胶束法得到的蛋白质样品的粒径分布有两个窄峰,而碱溶酸沉法得到的蛋白溶液只有一个宽峰,位置在反胶束法样品的两个峰中间。表明碱溶酸沉法得到的大豆分离蛋白溶液中分子间聚集现象整体比反胶束方法明显。这和表面疏水性的结果是一致的。
2.5 蛋白质的热稳定性
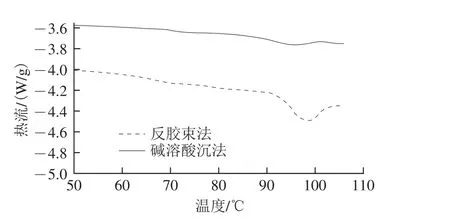
图4 两种方法得到的大豆分离蛋白的热变性结果Fig. 4 Differential scanning calorimetry thermograms of different SPIs
从图4中可以看出,两种方法得到的大豆分离蛋白都有两个吸热峰,这主要是由于大豆分离蛋白有7S和11S两种亚基。反胶束法对应的变性峰值分别为(70.03±0.56)℃和(98.85±0.67)℃;传统碱溶酸沉法对应的变性峰值分别为(75.12±0.84)℃和(97.37±0.16)℃。因此,在70~75 ℃时蛋白已经开始变性,这很有可能是部分7S亚基发生变性导致的,且反胶束法对应的变性峰温度较低,说明反胶束法得到的7S亚基热稳定性较差;而97~98 ℃时的变性峰则为11S亚基发生变性导致的,这和Zhao Xiaoyan等[13]的结果一致。不同方法得到的大豆分离蛋白的两个变性峰的差异主要是因为7S和11S亚基含量存在差异[30];此外,也有可能是蛋白质的二级结构或部分亚基发生改变导致的[31]。反胶束法得到的大豆分离蛋白吸热峰的焓变为(6.06±0.58)J/g和(0.31±0.02)J/g;碱溶酸沉法对应的焓变为(8.47±0.60)J/g和(0.84±0.34)J/g。两种方法焓变的差异和前述结构方面的变化一致,即碱溶酸沉法得到的大豆分离蛋白结构上已经部分展开,发生变性聚集时所需要的能量更高。因此,从变性温度上可以看出,反胶束法得到的大豆分离蛋白的变性温度较高,但从焓变可以看出,其达到变性温度时其所需的热量并不大,更易受到热破坏[17]。
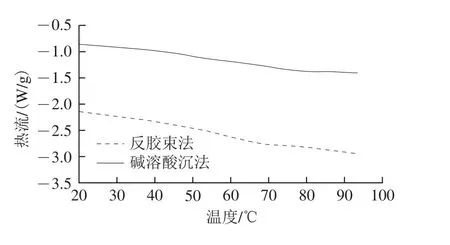
图5 两种方法得到的大豆分离蛋白的相态转变结果Fig. 5 Glass transition temperature of different SPIs
从图5中可以看出,反胶束法得到的大豆分离蛋白的玻璃态转变温度有两个,分别是(47.76±1.62)℃和(69.38±0.62)℃;而碱溶酸沉法得到的大豆分离蛋白只出现了一个较为明显的玻璃态转变温度((58.81±1.42)℃)。结合图4可以推测,反胶束法得到的大豆分离蛋白的两个玻璃态转变温度分别对应的依然是7S和11S亚基;而碱溶酸沉法分离得到的大豆分离蛋白只出现了一个玻璃态转变温度,有可能是两种成分玻璃态转变出现叠加的结果。反胶束法得到的蛋白质的玻璃态转变温度略低于碱溶酸沉法。
2.6 蛋白质的凝胶特性
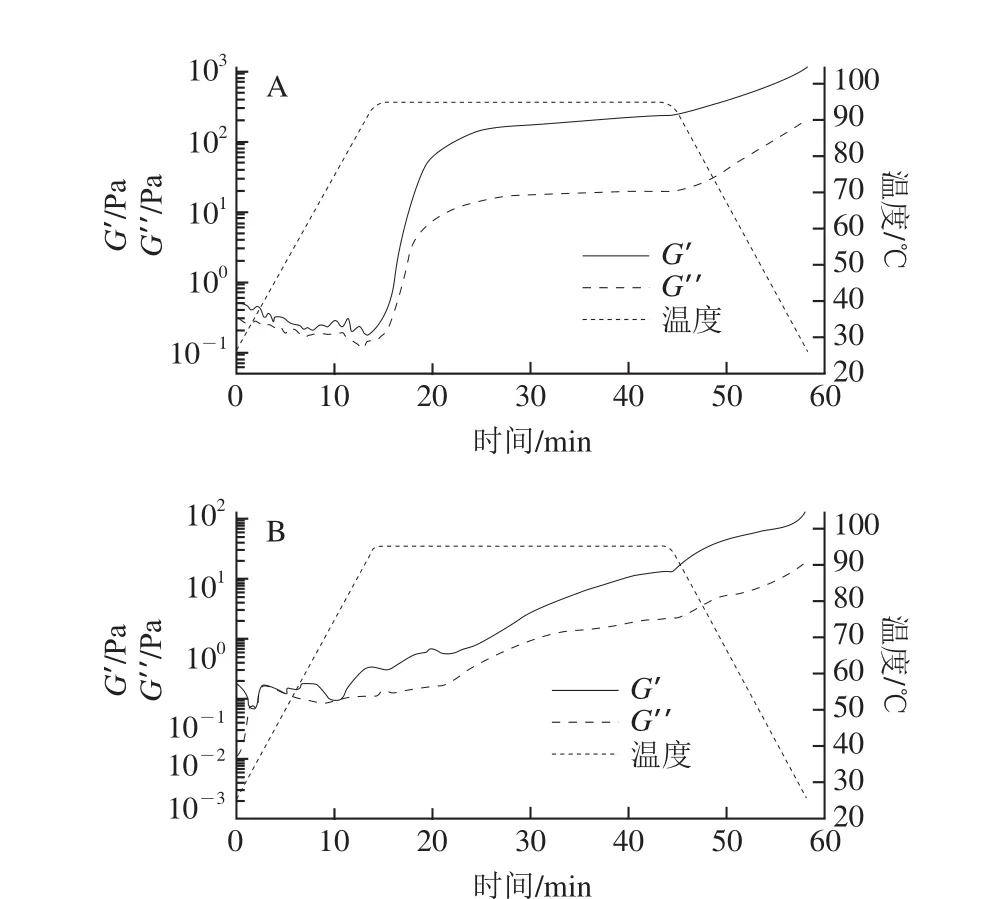
图6 碱溶酸沉法(A)和反胶束法(B)制得大豆分离蛋白凝胶的温度扫描图Fig. 6 Temperature curves of SPIs of traditional alkali dissolution and acid precipitation (A) and reverse micelle (B)
对于未知样品进行动态流变学测定前都要预先进行应力扫描确定动态黏弹区[32]。确定线性黏弹区后,最终选定温度扫描的条件为0.5 Hz、1%形变。如图6所示,从整体趋势上可以看出两种蛋白在加热过程中G′和G”同时上升,且G′高于G”,因此可以确定凝胶体系的形成。由于两种样品均在相同的条件下加热,因此G′开始激增的时间先后可以作为成胶能力的衡量标准[33]。碱溶酸沉法得到的大豆分离蛋白开始形成凝胶的温度点为95.00 ℃,而反胶束法对应的大豆分离蛋白开始形成凝胶的温度点为78.44 ℃,可以明显看出反胶束法相较于传统碱溶酸沉法而言更易形成凝胶。但从图6A中可以看出,G′和G”在极短的时间内就大幅度上升;而图6B中反胶束法得到的大豆分离蛋白的G′和G”却整体呈现缓慢的上升趋势,慢慢形成凝胶网络结构,即反胶束法得到的大豆分离蛋白凝胶形成的速度较慢。此外,两种样品在温度扫描结束时分别对应的G′数值可以作为凝胶刚性的表征[34]:图6A中碱溶酸沉法得到的大豆分离蛋白对应的G′为1 108.96 Pa;图6B中反胶束法得到的大豆分离蛋白对应的G′为122.70 Pa。因此,碱溶酸沉法得到的大豆分离蛋白形成的凝胶强度远优于反胶束法。这可能是由于反胶束法得到的大豆分离蛋白的巯基和二硫键的含量较低[13,35],致使其无法形成较为紧密的凝胶体系。
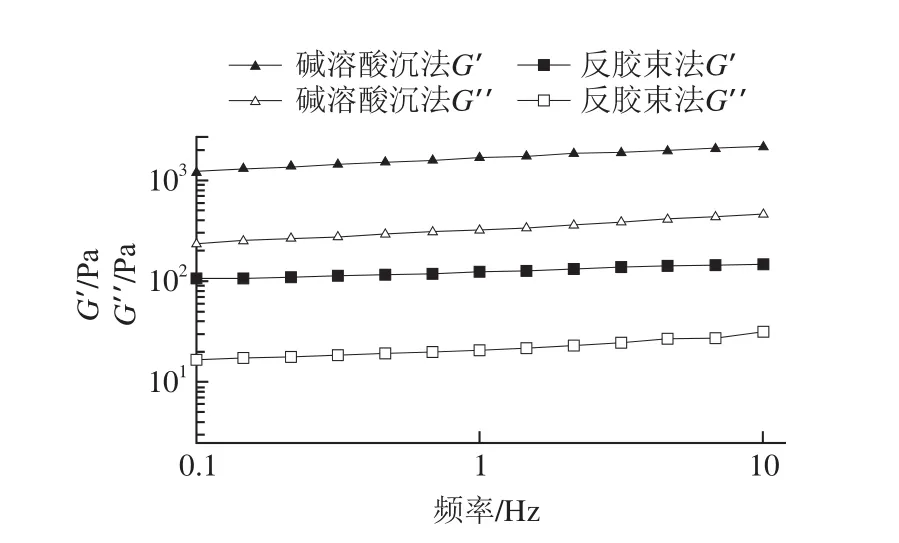
图7 两种方法得到的大豆分离蛋白凝胶的频率扫描图Fig. 7 Frequency curves of different SPIs
此外,还可以通过G′和G”以及它们的频率依赖性(频率扫描图)来研究蛋白样品所形成的凝胶的状态,典型凝胶的G′和G”是相互平行的,且G′>G”[36]。从图7中可以看出,两种样品的G′和G”是相互平行的,且G′>G”,即形成了典型的凝胶结构。Arzeni等[35]报道频率扫描曲线的斜率与凝胶中聚集物的尺寸有关,斜率越大说明凝胶中分子的聚集程度越大。因此,对照图7可以看出,碱溶酸沉法得到的蛋白凝胶的频率扫描曲线斜率较大,即蛋白质在形成凝胶时分子交联现象更多,这也极有可能是由该方法得到的蛋白质中二硫键含量的差异引起的,因为分子间的二硫键能促使蛋白分子亚基间形成束状结构单元,进而形成网络结构[37]。
3 结 论
结构上可以看出,反胶束方法得到的大豆分离蛋白的β-折叠结构含量较少,而β-转角结构含量较多,表面疏水性较低,表明反胶束环境相较于碱溶酸沉法能最大程度地保持蛋白质的分子结构。分析蛋白质的热力学特性可以得出结论,不同方法得到的大豆分离蛋白的热力学变化均呈现两个变性峰,分别对应两种主要亚基(7S和11S球蛋白)。反胶束方法得到的大豆分离蛋白的主成分7S亚基对应的变性温度较低,而11S亚基的变性温度较高,但变性过程中焓变低于碱溶酸沉法,即达到变性温度时反胶束法对应的蛋白质更易变性;且反胶束法得到的大豆分离蛋白具有较低的玻璃态转变温度。对两种蛋白样品形成凝胶的过程进行动态流变学实验,发现反胶束法对应的蛋白质在较低温度处G′和G”就开始发生变化,这是因为反胶束法对应的蛋白质起始变性温度较低,且凝胶形成速度较慢,最终形成的凝胶体系强度也低于碱溶酸沉法得到的大豆分离蛋白。综上所述,反胶束法相较于碱溶酸沉法而言,能较好地保持蛋白质的分子结构,但耐热性较差,因此在较低温度下就开始形成凝胶,但最终所成凝胶的强度较低。
猜你喜欢
中国造纸(2022年9期)2022-11-25
中国造纸(2022年8期)2022-11-24
今日农业(2022年16期)2022-11-09
中国化肥信息(2022年5期)2022-08-30
今日农业(2021年20期)2021-11-26
今日农业(2021年14期)2021-10-14
陶瓷学报(2021年1期)2021-04-13
军事文摘(2020年20期)2020-11-16
中学生数理化·八年级物理人教版(2020年12期)2020-01-01
环球时报(2019-04-03)2019-04-03
