
海藻糖合酶在枯草芽孢杆菌中的高效表达
2019-05-07 04:51王希晖刘洪玲隋松森杨少杰王瑞明王腾飞
食品与发酵工业 2019年7期
王希晖,刘洪玲,隋松森,杨少杰,王瑞明,王腾飞*
1(生物基材料与绿色造纸国家重点实验室(LBMP)(齐鲁工业大学),山东 济南,250353) 2(山东省微生物工程重点实验室(齐鲁工业大学),山东 济南,250353) 3(诸城东晓生物科技有限公司,山东 潍坊,261000)
海藻糖[1]是由2个葡萄糖分子通过1,1-糖苷键结合而成的非还原性糖,因其特殊的生物学功能和独特的应用价值,广泛应用于食品、药品、农牧业等行业,其发展前景非常广阔,但生产上的不足导致其应用受到一定程度的限制。当前获得认可的海藻糖生产工艺是利用海藻糖合酶(trehalose synthase)[2]直接转化麦芽糖生成海藻糖,但海藻糖合酶的获取受到限制,影响了海藻糖的生产。
因此,为实现海藻糖合成酶的安全高效表达,安全的宿主菌和高效的转录表达系统的选择是关键。枯草芽孢杆菌(Bacillussubtilis)作为一种革兰氏阳性菌[3],由于其自身的非致病特性和分泌外源蛋白能力强的特性,以及有着长期制备发酵食品的历史,已是公认的一种安全菌株。因此本试验采用B.subtilisWB800n进行宿主表达,可解决宿主菌自身产内毒素的不安全因素。枯草芽孢杆菌有多种不同的转录表达系统[4],根据启动子诱导机制的不同,可分为4种类型,分别是诱导型[5]、自诱导型[6]、时期特异型[7]和组成型[8]启动子,由于诱导型启动子是在发酵过程中添加化合物诱导或受环境变化来控制基因转录,发酵条件受到限制,另外3种转录方式都是非诱导型,发酵过程简便高效。
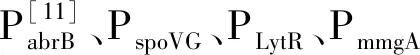
1 材料与方法
1.1 材料
1.1.1 菌株与质粒
恶臭假单胞杆菌KT2440(PseudomonasputidaKT2440)、大肠杆菌DH5α(E.coliDH5α)、枯草芽孢杆菌WB800n (B.subtilisWB800n)菌株,质粒是穿梭表达质粒pHT01。
1.1.2 实验试剂
pZERO-Blunt零背景平末端快速连接试剂盒、2×Phanta Max Master Mix、2×TaqPCR Master Mix:艾德莱生物科技有限公司;T4 DNA连接酶、限制内切酶:Thermo公司;SanPrep柱式DNA胶回收试剂盒、DNA纯化回收试剂盒:生工生物工程(上海)股份有限公司。
1.1.3 引物及核苷酸序列
引物通过Oligo 7.0软件设计,由上海生工生物工程公司合成,其引物序列见表1。测序引物为T7启动子的通用引物。

表1 质粒构建用引物
1.1.4 培养基
LB培养基:NaCl 10 g/L、蛋白胨10 g/L、酵母浸粉5 g/L,pH 7.0;
LB固体培养基:在LB培养基中加入质量浓度20 g/L琼脂粉;
TB培养基:胰蛋白胨12 g/L、酵母浸粉24 g/L、甘油4 mL/L、KH2PO417 mmol/L、K2HPO472 mmol/L;
优化后TB培养基:胰蛋白胨12 g/L、酵母浸粉24 g/L、蔗糖12g/L、KH2PO46.5 mmol/L、K2HPO427 mmol/L;
GM增殖培养基:NaCl 10 g/L、蛋白胨10 g/L、酵母浸粉5 g/L、山梨醇0.5 mol/L;
RM复苏培养基:NaCl 10 g/L、蛋白胨10 g/L、酵母浸粉5 g/L、山梨醇0.5 mol/L、甘露醇0.38 mol/L;
ETM电转缓冲液:山梨醇0.5 mol/L、甘露醇0.5 mol/L、海藻糖0.5 mol/L、甘油体积分数10%。
1.2 方法
1.2.1 穿梭表达载体的构建
1.2.1.1 PCR获得目的基因片段
根据NCBI上报道的PsrfA和4个时期特异性启动子PabrB、PspoVG、PLytR、PmmgA的基因序列人工合成PsrfA和PabrB-spoVG-LytR-mmgA的基因片段,并对启动子PsrfA进行了密码子优化[14],将PsrfA启动子的-35区(GTGATA)和-10区(TAAACT)分别改为-35区(TTGACT)和-10区(TATAAT)。
1.2.1.2 pHT01-PsrfA- treS表达载体的构建
以人工合成的PsrfA基因序列为模板,用引物SrfA-F/SrfA-R通过PCR扩增得到长度为606 bp的PsrfA基因片段,以恶臭假单胞杆菌KT2440基因组为模板,用引物TreS-1-F/TreS-R经过PCR扩增得到长度为2 067 bp的treS基因片段,再通过重叠PCR的方法将2个片段扩增连接得到长度为2 673 bp的带有His标签的PsrfA-treS基因片段。将片段连接到pZERO-Blunt平末端载体,送测序公司进行基因测序,测序正确后,采用限制性内切酶BamH I/AatII对pZERO-PsrfA-treS和pHT01双酶切,再用T4连接酶连接得到pHT01-PsrfA-treS重组载体。载体构建流程如图1所示。
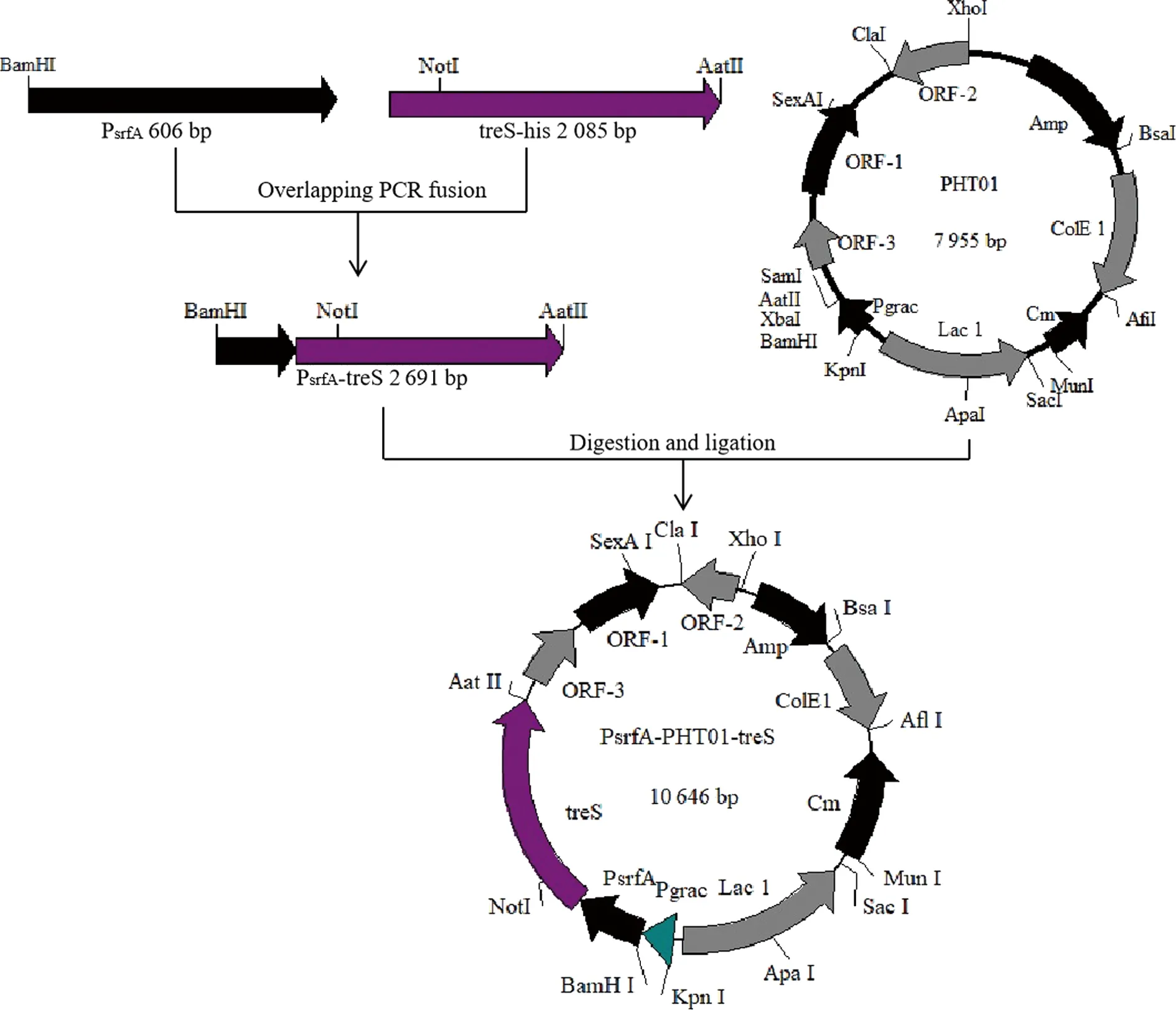
图1 PHT01-PsrfA-TreS质粒载体的构建
Fig.1 Construction of pHT01-PsrfA-treS plasmid vector
1.2.1.3 pHT01-P43-treS表达载体的构建
以枯草芽孢杆菌基因组为模板,用引物P43-F/P43-TreS-R PCR扩增得到长度445 bp的P43基因片段,在treS基因315 bp处有酶切位点NotI,以构建的pHT01-PsrfA-treS为模板,用引物TreS-P43-F/TreS-1-R PCR扩增得到长度为315 bp的treS-1基因片段,再通过重叠PCR的方法将2个片段连接扩增得到长度为760 bp的P43-treS-1基因片段。将片段连接到pZERO-Blunt平末端载体送到测序公司进行基因测序,测序正确后,用限制性内切酶BamH I/NotI对pZERO-P43-treS-1和pHT01-PsrfA-treS进行双酶切,再用T4连接酶连接得到pHT01-P43-treS重组载体。
1.2.1.4 pHT01-PabrB-spoVG-LytR-mmgA-treS表达载体的构建
依据NCBI中公布的启动子序列,对PabrB-spoVG-LytR-mmgA基因序列进行人工合成后,用引物PabrB-F/PmmgA-TreS-R PCR扩增得到长度1 200 bp的PabrB-spoVG-LytR-mmgA基因片段,在treS基因315 bp处有酶切位点NotI,以构建的pHT01-PsrfA- treS为模板,用引物TreS-mmgA-F/TreS-1-R PCR扩增得到长度为315 bp的treS-1基因片段,再通过重叠PCR的方法将2个片段连接扩增得到长度为1 515 bp的PabrB-spoVG-LytR-mmgA-treS-1基因片段。将片段连接到pZERO-Blunt平末端载体送到测序公司进行基因测序,测序正确后,限制性内切酶BamH I/NotI对pZERO-PabrB-spoVG-LytR-mmgA-treS-1和载体pHT01-PsrfA-treS进行双酶切,再用T4连接酶连接得到pHT01- PabrB-spoVG-LytR-mmgA-treS重组载体。
1.2.2 产海藻糖合酶重组枯草芽孢杆菌的构建
1.2.2.1 枯草芽孢杆菌感受态的制备
挑取新鲜的LB固体培养基(添加新霉素抗性)表面的B.subtilisWB800n单菌落,接种于5 mL的LB培养基中,37 ℃、200 r/min培养过夜;取500 μL上述菌液转接到50 mL GM增殖培养基中,37 ℃、200 r/min培养至OD600=1.0;将菌液转移至已灭过菌的100 mL离心管,冰水浴10 min后,5 000 r/min、4 ℃离心10 min,收集菌体;用预冷的电转缓冲液ETM洗涤3~4次,将洗涤后的菌体重悬于500 μL的ETM中,即为枯草芽孢杆菌电转化感受态细胞[15];将制备好的感受态细胞分装为每管60 μL,-80 ℃保存备用。
1.2.2.2 构建重组菌B.subtilisWB800n(pHT01-PsrfA-treS、pHT01-P43-treS和pHT01-PabrB-spoVG-LytR-mmgA-treS)
从-80 ℃冰箱取出3管60 μL的感受态细胞(新制备可直接用,不需要放置冰箱),分别加入6 μL的质粒pHT01-PsrfA-treS、pHT01-P43-treS和pHT01-PabrB-spoVG-LytR-mmgA-treS混合均匀,预冷5 min后将混合的菌液加入2 mm的电转杯(电转杯需要提前处理:无水乙醇浸泡10 min,然后用吹风机吹干,置于无菌操作台紫外照射20 min,然后盖上盖子,放置冰中预冷)中,用Eppendorf电转仪在2 000 V、5 ms条件下电击1次。电转完毕后,迅速加入1 mL RM复苏培养基,37 ℃、200 r/min复苏4 h后,离心重悬后涂布在含氯霉素(40 μg/mL)固体LB培养基,放在37 ℃恒温培养箱中倒置培养1~2 d,筛选抗氯霉素的菌株。
1.2.2.3 验证重组菌
挑取上述抗氯霉素菌株的单菌落作为模板,分别通过上下游引物来进行PCR扩增,扩增产物利用1%琼脂糖凝胶电泳进行验证,最终获得阳性克隆菌株。
1.3 摇瓶培养条件的优化
(1)碳源优化。分别以可溶性淀粉、麸皮、葡萄糖、麦芽糖、海藻糖、乳糖、蔗糖为碳源,进行摇瓶发酵,TB培养基做对照。
(2)碳氮比优化。胰蛋白胨与酵母浸粉的比例分别选择0∶36、6∶30、12∶24、18∶18、24∶12、30∶6、36∶0,碳源使用优化后的蔗糖,进行摇瓶发酵,TB培养基做对照。
(3)不同发酵温度对发酵结果的影响。使用优化碳源和氮源的培养基进行摇瓶发酵,温度分别设置30、33、37、40和45 ℃,验证发酵温度对酶活的影响。
(4)培养基初始pH值优化。以优化后的TB培养基作为基本培养基,改变培养基中K2HPO4和KH2PO4的比例,分别调节培养基初始pH值为6.0、6.5、7.0、7.5、8.0、8.5,进行摇瓶发酵。
1.4 5 L发酵罐发酵培养
使用总体积为5 L发酵罐(BLBIO-5GJ)[16],工作体积为3 L,发酵过程中,温度维持37 ℃,pH值维持在8.0,(pH值通过滴加10%的氨水来维持),并且通过对通风量与搅拌速率的控制使溶氧(dissolved oxygen,DO)维持在20%~40%。初始培养基为优化后的TB培养基,另加入40 μg/mL的氯霉素。发酵时间32 h,每隔10、13、16、20、24、28、32 h取样1次,直至发酵结束。
1.4.1 酶活测定
将在不同发酵时间取出的40 mL菌样于8 000 r/min离心10 min,弃去上清得到沉淀,用配制好的10 mmol/L pH 8.0 PBS缓冲液重悬,经超声波破碎得到粗酶液。取5 mL粗酶液与5 mL 60%的麦芽糖溶液混匀,在25 ℃水浴锅中转化4 h后,于100 ℃煮沸10 min,灭活,通过液相色谱分析仪测定其转化的海藻糖含量,并计算出不同发酵时间的每克菌体的酶活。
酶活力单位[17]定义:在酶的最佳酶反应条件下(25 ℃,pH 8.0),将质量浓度300 g/L麦芽糖转化为海藻糖,每小时产生1 μmol海藻糖所需的酶量定义为1个酶活单位(U)。
1.4.2 SDS-PAGE检测分析
取不同培养条件的发酵液,12 000 r/min离心10 min,弃去上清,用0.05 mol/L pH 8.0的PBS缓冲液重悬,超声破碎,加入SDS-PAGE样品缓冲液后煮沸10 min,样品进行SDS-PAGE(分离胶浓度为12%,浓缩胶浓度为5%)。电泳结束后凝胶染色4 h然后脱色,将脱色后的凝胶用凝胶成像系统观察。
2 结果与分析
2.1 重组菌的构建
2.1.1 重组质粒pHT01-PsrfA-treS、pHT01-P43-treS和pHT01-PabrB-spoVG-LytR-mmgA-treS的构建
通过PCR和重叠PCR技术得到PsrfA-TreS、P43-TreS-1和PabrB-spoVG-LytR-mmgA-TreS-1基因片段,通过1%琼脂糖凝胶电泳检测发现,在2 700、750和1 500 bp左右出现特异性电泳条带,如图2-a、b、c所示,与理论值2 673、760、和1 515 bp相符,表明获得了PsrfA-TreS、P43-TreS-1和PabrB-spoVG-LytR-mmgA-TreS-1基因片段。将制得的片段连接到表达载体pHT01相应酶切位点,重组载体pHT01-PsrfA-TreS通过限制性内切酶BamH I/AatII对载体进行酶切验证,载体pHT01-P43-treS和pHT01-PabrB-spoVG-LytR-mmgA-treS通过限制性内切酶BamH I/NotI对载体进行酶切验证,用1%琼脂糖凝胶电泳检测发现,在2 700、750和1 500 bp左右出现特异性电泳条带,如图2-d、e、f所示,表明表达载体pHT01-PsrfA-treS、pHT01-P43-treS和pHT01-PabrB-spoVG-LytR-mmgA-treS构建成功。
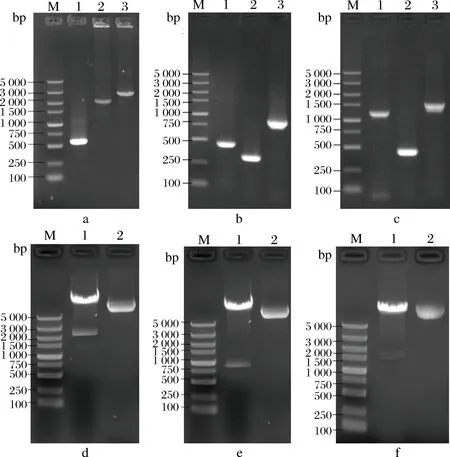
a:M-Maker DL5000;1-PsrfAband;2-treS band;3-PsrfA-treS band;b:M-Maker DL5000;1-P43band;2-treS-1 band;3-P43-treS-1 band;c:M-Maker DL5000;1-PabrB-spoVG-LytR-mmgAband;2-treS-1 band;3-PabrB-spoVG-LytR-mmgA-treS-1 band;d:M-Maker DL5000; 1-Double enzyme digestion of pHT01-PsrfA-treS; 2-pHT01-PsrfA-treS;e:M-Maker DL5000; 1-Double enzyme digestion of pHT01-P43-treS; 2-pHT01-PsrfA-treS;f:M-Maker DL5000; 1-Double enzyme digestion of pHT01-PabrB-spoVG-LytR-mmgA-treS; 2-pHT01- PabrB-spoVG-LytR-mmgA-treS图2 基因的PCR扩增电泳图和重组表达质粒的酶切验证图
Fig.2 PCR amplified electrophoresis and restrictionenzyme digestion identifieation of recombinant plasmid
2.1.2 重组菌B.subtilisWB800n/pHT01-PsrfA-treS、B.subtilisWB800n/pHT01-P43-treS和B.subtilisWB800n/pHT01-PabrB-spoVG-LytR-mmgA-treS的构建
提取质粒pHT01-PsrfA-treS、pHT01-P43-treS和pHT01-PabrB-spoVG-LytR-mmgA-treS,并用核酸超微量分光光度计(BioFuture MD2000)测DNA质量浓度,结果显示,DNA质量浓度为220 ng/μL左右。将6 μL提取的质粒与60 μLB.subtilisWB800n感受态细胞[18]混合并进行电转化,分别使用上下游引物SrfA-F/TreS-1-R、P43-F/TreS-1-R和PabrB-F/TreS-1-R对阳性克隆子进行PCR验证,通过1%琼脂糖凝胶电泳检测发现,在1 000、750和1 500 bp左右出现特异性电泳条带,如图3-a、b、c所示,与理论值921、760、和1 515 bp相符,表明质粒载体pHT01-PsrfA-treS、pHT01-P43-treS和pHT01-PabrB-spoVG-LytR-mmgA-treS成功转化到B.subtilisWB800n中,并获得了阳性克隆菌株。
a: M-Maker AL5000;1-positive control plasmid pHT01-PsrfA-treS; 2-negative control B.subtilis WB800n; 3~8-PsrfA-treS-1 fragment electrophoretic diagram;b: M-Maker AL5000;1-positive control plasmid pHT01-P43-treS; 2-negative control B.subtilis WB800n; 3~8-P43-treS-1 fragment electrophoretic diagram;c: M-Maker AL5000;1-positive control plasmid pHT01-PabrB-spoVG-LytR-mmgA-treS; 2-negative control B.subtilis WB800n; 3~8-PabrB-spoVG-LytR-mmgA-treS-1 fragment electrophoretic diagram图3 重组菌的菌落PCR验证图
Fig.3 Colony PCR validation of recombinant bacterial
2.2 启动子的筛选
将构建成功的重组菌枯草芽孢杆菌B.subtilisWB800n/pHT01-PsrfA-treS、B.subtilisWB800n/pHT01-P43-treS和B.subtilisWB800n/pHT01-PabrB-spoVG-LytR-mmgA-treS分别接种到LB培养基中,发酵培养分析不同启动子重组菌的单位菌体酶活,结果如图4所示,3个重组菌均能在胞内表达海藻糖合酶,而且随着发酵时间的延长,单位菌体酶活呈现先增加后降低的趋势。发酵过程中组成型启动子P43启动子表现出较高的酶活,其次是4个时期特异性串联启动子PabrB-spoVG-LytR-mmgA,最后是PsrfA启动子。P43启动子表达的酶活最高达到约2 167 U/g,这说明P43启动子的启动强度最大,较不同时期特异性启动子串联特性强,而PsrfA启动子是群体密度依赖型启动子[19],其表达酶活的时间延迟,这主要是因为在密度低时,启动子的启动强度受到限制。
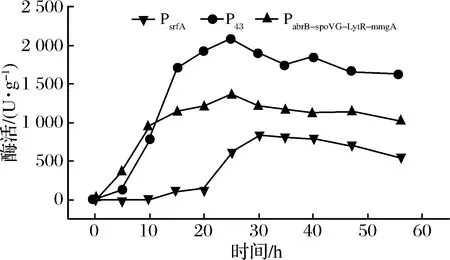
图4 胞内表达TreS重组菌LB培养基酶活
Fig.4 Intracellular TreS enzyme activity of recombinant bacteria in LB medium
为了进一步验证3个启动子的酶活特性,将3个重组菌接种至更利于外源酶表达的TB培养基中进行发酵培养,结果如图5所示。菌体表达的总酶活均有明显增加,P43调控的重组菌最高酶活达到3 481 U/g,但3个启动子的表达酶活依然遵循LB培养基相同的情况,亦在发酵的后期,酶活下降迅速,这主要由2个原因造成,一是后期菌体为了维持生长,对表达的外源酶进行降解;二是发酵后期,因为营养的逐渐匮乏,多数菌体产生芽孢,造成酶的降解。
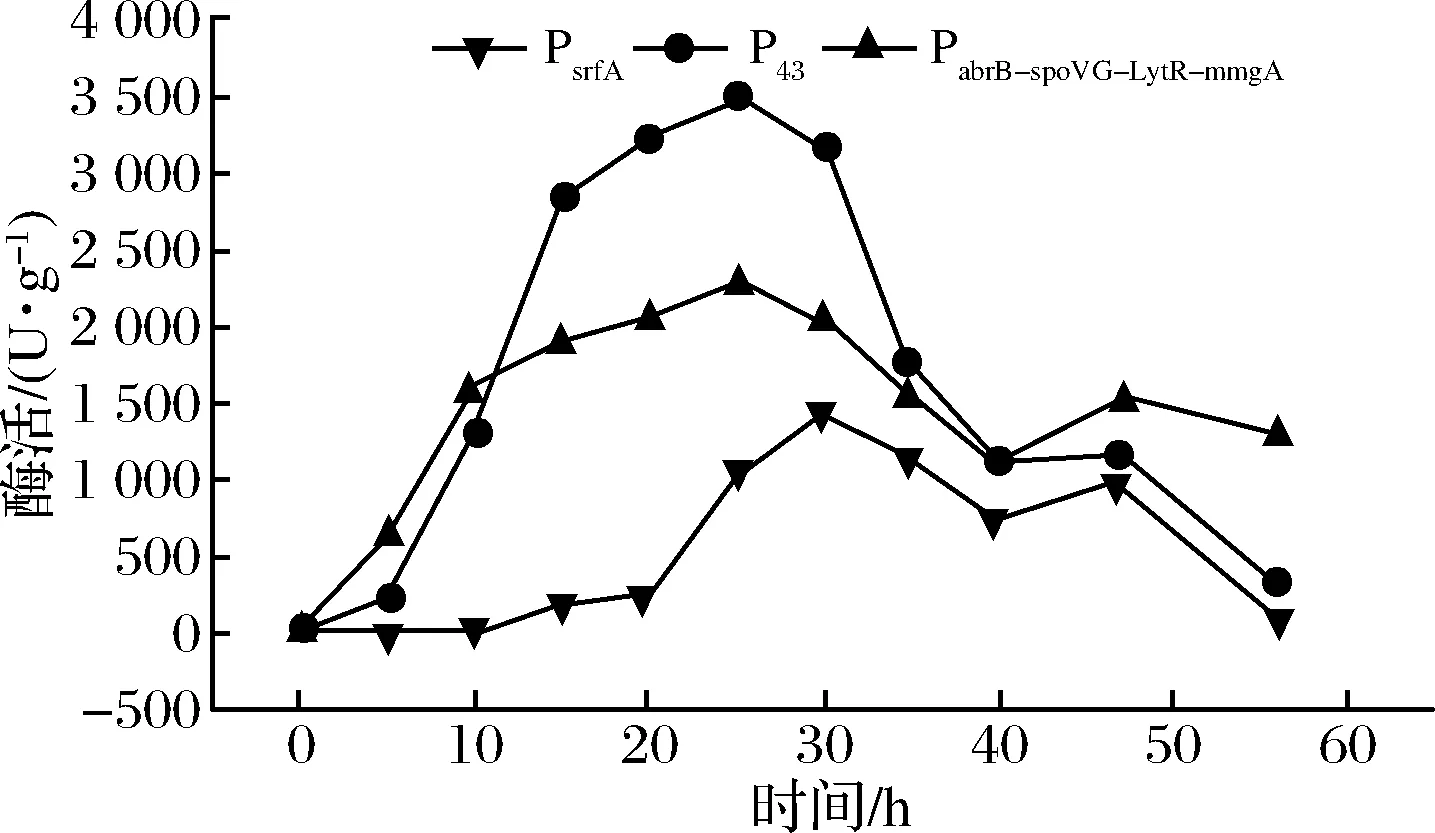
图5 胞内表达TreS重组菌TB培养基酶活
Fig.5 Intracellular TreS enzyme activity of recombinant bacteria in TB medium
2.3 摇瓶发酵条件优化
2.3.1 碳源优化
以TB培养基作为基本培养基[20],改变TB培养基中的碳源组分,将重组菌B.subtilisWB800n/pHT01-P43-treS在500 mL含有100 mL不同碳源的TB培养基中进行发酵酶活分析。在37 ℃,200 r/min下摇瓶发酵培养24 h。分析不同碳源对发酵酶活的影响,从图6、图7可以看出,不同碳源对发酵酶活有不同的影响,以蔗糖为碳源时酶活达到5 763 U/g,明显高于其他碳源。对蔗糖的添加量进行了优化,最终得出,添加12 g/L蔗糖酶活最高。
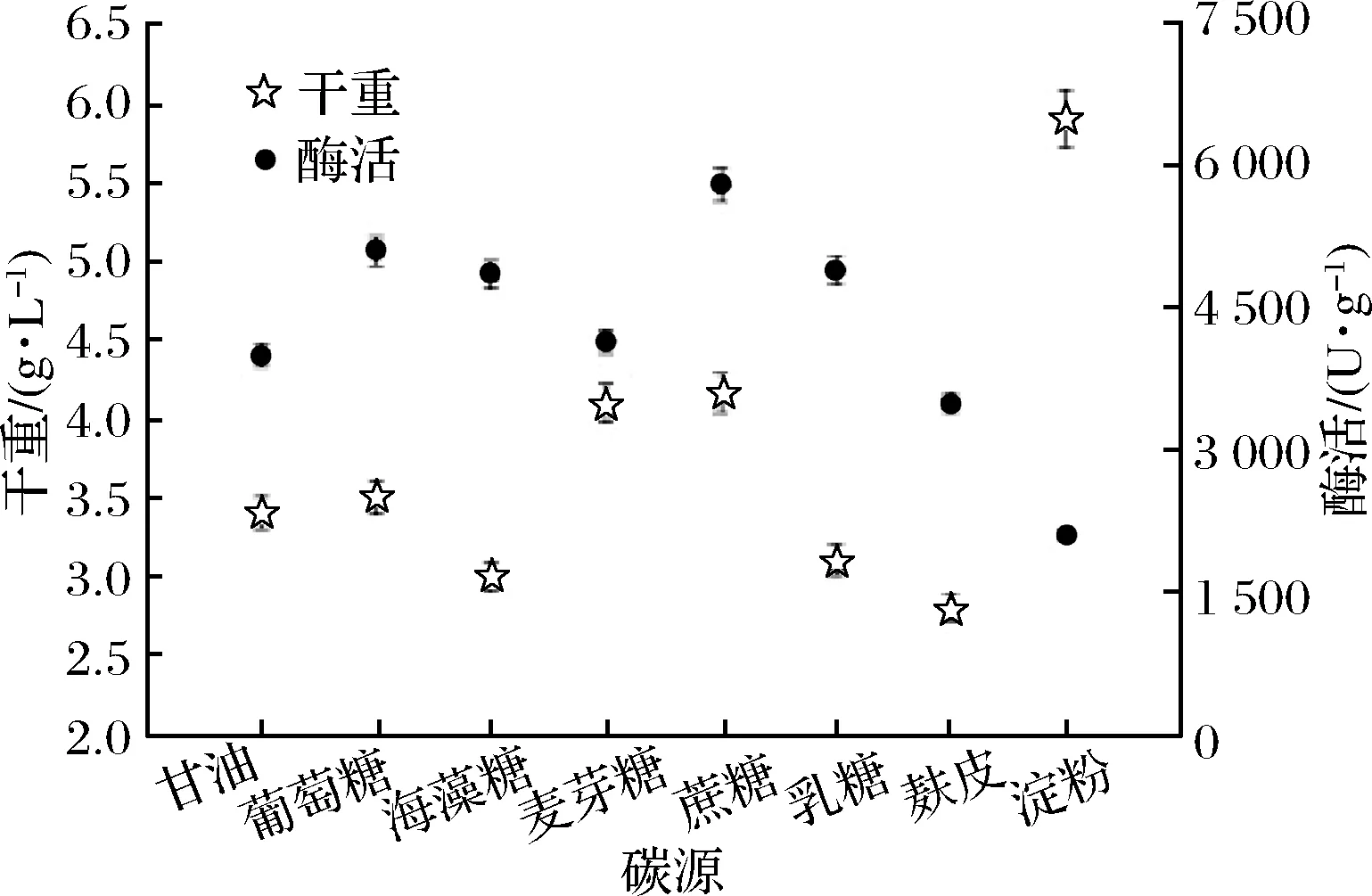
图6 B.subtilis WB800n/pHT01-P43-treS的不同碳源比较
Fig.6 Different carbon source comparison of B.subtilis WB800n/pHT01-P43-treS
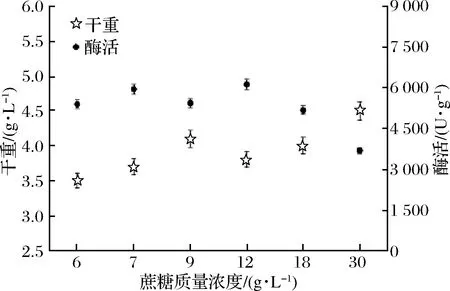
图7 B.subtilis WB800n/pHT01-P43- treS的不同蔗糖质量浓度比较
Fig.7 Different sucrose concentration comparison of B.subtilis WB800n/pHT01-P43- treS
2.3.2 氮源优化
氮源是微生物生长的必要元素,同时在发酵过程中也是增加成本的因素之一[21],因此,有必要分析氮源对发酵酶活的影响。在上述优化后的TB培养基中,改变其中的胰蛋白胨与酵母浸粉的比例,在37 ℃,200 r/min下摇瓶发酵培养24 h,验证不同比例的2种氮源对发酵液酶活的影响,如图8所示,原有的胰蛋白胨∶酵母浸粉比例为12∶24时对酶活的影响具有明显的优势,因此维持原有的比例成分不变。

图8 B.subtilis WB800n/pHT01-P43- treS的不同氮源比例的比较
Fig.8 Different nitrogen sources ratio comparison of B.subtilis WB800n/pHT01-P43- treS
2.3.3 发酵温度对酶活的影响
对原始TB培养基的主要成分进行适当优化后,进一步分析发酵温度对产酶的影响。重组菌先在37 ℃,200 r/min条件下培养8 h,然后改变培养温度。从图9可以看出,37 ℃为最佳温度,温度过高过低都不利于海藻糖合酶的表达。
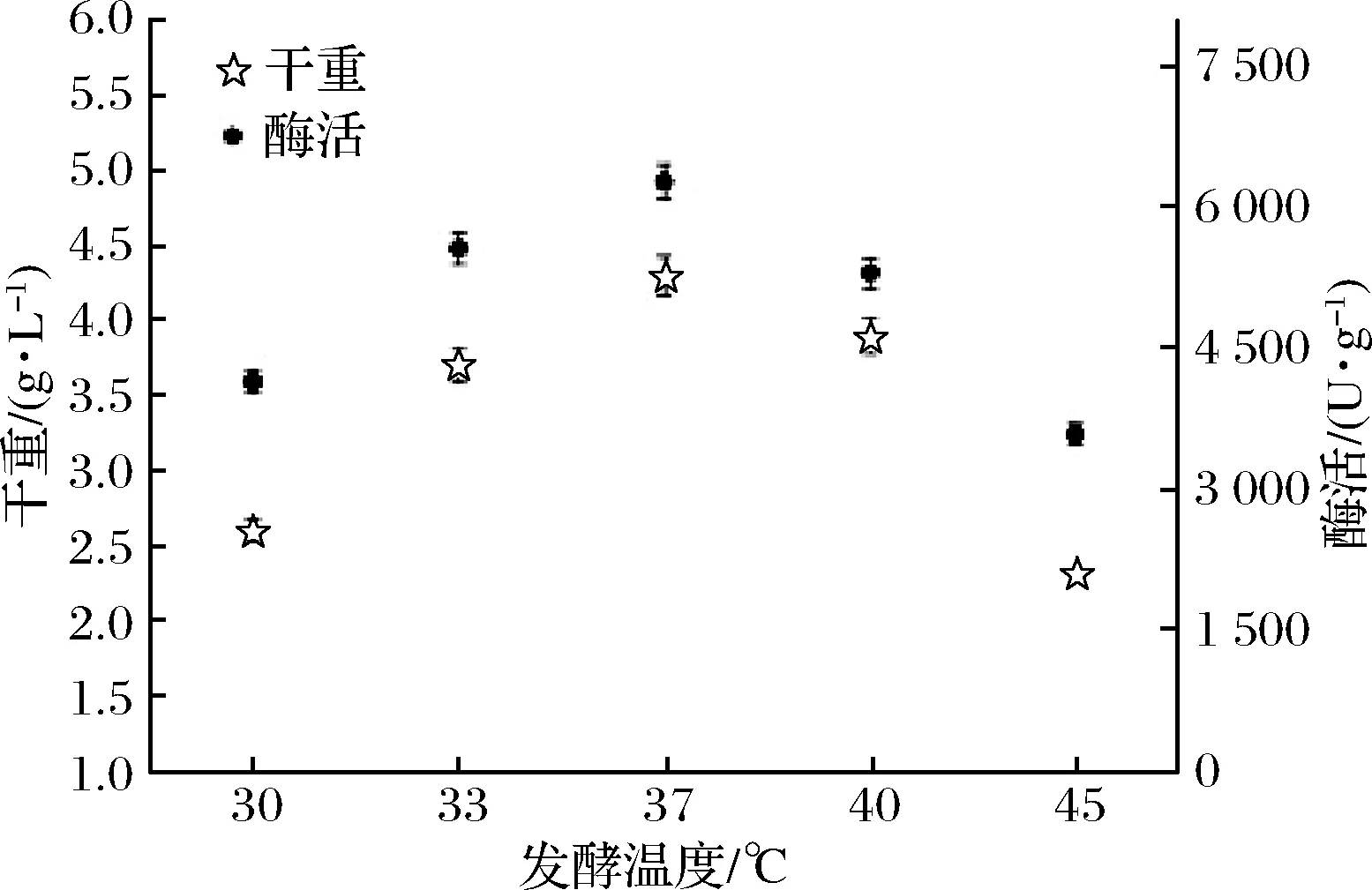
图9 B.subtilis WB800n/pHT01-P43-treS的发酵温度的比较
Fig.9 Different fermentation temperatures comparison of B.subtilis WB800n/pHT01-P43-treS
2.3.4 发酵pH值对产酶的影响
对培养基的主要成分进行适当优化后,进一步分析不同发酵pH值对产酶的影响,以不同pH值的磷酸盐缓冲溶液配制发酵培养基,在37 ℃,200 r/min下摇瓶发酵培养24 h,发酵液酶活如图10所示。
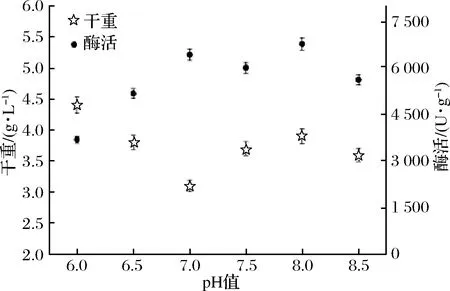
图10 B.subtilis WB800n/pHT01-P43-treS的发酵pH值的比较
Fig.10 Different fermentation pH comparison of B.subtilis WB800n/pHT01-P43-treS
结果表明,在pH 7.0~8.0总酶活相对较高,pH 8.0时,总酶活最高,且此时的菌体干重亦有明显优势。
2.4 发酵罐放大发酵培养
为进一步探讨产酶稳定性,将重组菌B.subtilisWB800n/pHT01-PsrfA-treS、B.subtilisWB800n/pHT01-P43-treS和B.subtilisWB800n/pHT01-PabrB-spoVG-LytR-mmgA-treS在装有优化后TB培养基的5 L发酵罐中进行发酵验证,发酵罐总装液量为3 L,转速500 r/min,通风量0.3 vvm,温度37 ℃,初始pH值7.0,发酵前8 h发酵液pH值缓慢上升至8.0,之后通过滴加NaOH维持pH值稳定在8.0。发酵结果如图11所示,结果表明,TreS的表达量仍然是P43>PabrB-spoVG-LytR-mmgA>PsrfA。
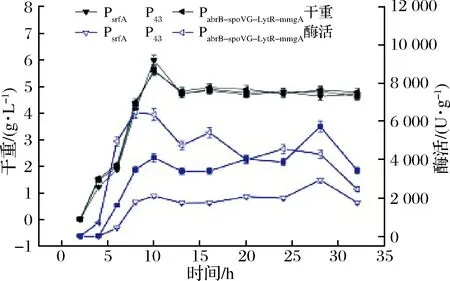
图11 胞内表达TreS重组菌的5 L发酵罐酶活
Fig.11 Enzyme activity of recombinant bacteria in 5 L fermentor
2.5 SDS -PAGE凝胶分析
不同发酵时间取出的样品经超声破碎后做蛋白电泳,由图12可以明显看出,在75 kDa处有明显的条带,与海藻糖合酶75.634 kDa理论值[22]相符,说明海藻糖合酶基因在重组菌中成功表达。
M-Marker;1-B.subtilis WB800n;2~9-10,13,16,20,24,28,32 h图12 B.subtilis WB800n/pHT01-P43-treS在5 L发酵罐中菌体破碎液SDS-PAGE分析
Fig.12 SDS-PAGE analysis of B.subtilis WB800n/pHT01-P43-treS in 5 L fermentor
3 结论
本研究基于海藻糖合成酶在生产上的不足导致其应用受到一定程度的限制,本试验通过基因改造的手段,利用枯草芽孢杆菌的非诱导表达系统构建了海藻糖合成酶在安全菌株枯草芽孢杆菌中的高效表达系统[23]。本研究分别将单启动子和多启动子与海藻糖合成酶基因融合后构建到大肠-枯草穿梭质粒pHT01[24]上,通过电转化方法转化到枯草芽孢杆菌感受态细胞中,得到了重组菌B.subtilisWB800n/pHT01-PsrfA-treS、B.subtilisWB800n/pHT01-P43-treS和B.subtilisWB800n/pHT01-PabrB-spoVG-LytR-mmgA-treS。通过利用基础LB、TB培养基初步验证重组菌的酶活,验证3种启动子的转录强度,筛选得到强度最高的启动子P43,得到高效表达海藻糖合酶的重组菌B.subtilisWB800n/pHT01-P43-treS,并对此重组菌进行发酵优化,进一步提高了海藻糖合酶的表达量,成功构建了自诱导表达海藻糖合成酶的安全高效表达系统,为后续制备食品级的酶制剂[25]奠定了基础。
猜你喜欢
环境工程技术学报(2022年3期)2022-06-05
作文周刊·小学四年级版(2022年8期)2022-03-11
中国土壤与肥料(2021年5期)2021-12-02
江西农业学报(2021年4期)2021-04-20
昆钢科技(2021年6期)2021-03-09
水生生物学报(2021年1期)2021-02-04
天津科技(2020年4期)2020-05-09
食品科学(2020年4期)2020-03-11
小学生必读(低年级版)(2019年5期)2019-08-30
食品与生活(2017年12期)2018-01-09
