
SLC12A3基因杂合突变致青少年Gitelman综合征
2019-05-22 03:08窦为娟王瑞风沈业琴
肾脏病与透析肾移植杂志 2019年2期
窦为娟 王瑞风 沈业琴 李 洁 杜 宏
病史摘要
现病史18岁男性患者,因“间断乏力3年,再发5d”于2018年8月入住我科。患者于2015年运动后出现乏力,伴有麻木、抽搐,于当地医院查血钾1.8 mmol/L,诊断为“低血钾”,后无明显诱因曾发作类似症状2次,近2年未系统诊治。半月前患者因不洁饮食出现腹泻,8~9次/d,持续4~5d,腹泻停止后1周于8月2日无明显诱因下出现四肢乏力,不能行走,查血钾1.8 mmol/L,血压正常(113/73 mmHg),予以静脉补钾治疗后患者仍有四肢乏力,复查血生化:钾2.3 mmol/L、镁0.5 mmol/L、钠142.4 mmol/L、氯88 mmol/L、钙2.57 mmol/L、总二氧化碳34.7 mmol/L,血气:pH 7.53、氧分压79 mmHg、二氧化碳分压43 mmHg、实际碳酸氢根36 mmol/L、碱剩余13.2 mmol/L,甲状腺功能、肾上腺皮质功能、生长激素、性激素、肾上腺CT平扫、心电图未见异常,病程中,患者无发热,无怕热多汗,无心慌胸闷,无恶心呕吐,无多饮多尿史,夜尿0~1次,饮食及睡眠可,体重无明显变化。
既往史否认服用利尿剂、缓泻剂及其他药物。个人史无特殊,未婚未育。
家族史患者父母体健,非近亲婚配,1妹体健,否认家族中同样疾病患者,否认家族性遗传病史。
体格检查体温36.4 ℃,血压113/73 mmHg,身高172 cm,体重50 kg,神志清楚,发育正常,双侧甲状腺不大,心、肺、腹查体无异常,无手足麻木感,Chvostek征及Trousseau征均阴性。
实验室检查
尿液 尿常规pH 7.5、比重1.010;尿量3 200 ml/24h,尿钾73.6 mmol/24h(血钾2.9 mmol/L),尿氯155.2 mmol/24h,尿钠150.72 mmol/24h,尿钙0.48 mmol/24h;肾小管功能:尿NAG17.7U/g·cr[参考值<9.7 U/(g·Cr)],RBP 1.4 mg/L(参考值<0.7 mg/L),中性粒细胞明胶酶相关脂质运载蛋白13.0 ng/ml(参考值0.9~100 ng/ml),尿溶菌酶测定0.52 mg/L(参考值<0.5 mg/L),尿C3 2.0 mg/L(参考值≤2.76 mg/L),尿α2-MG 2.0 mg/L(参考值≤2.76 mg/L)。
血液 血液钾2.7 mmol/L,钠139 mmol/L,镁0.64 mmol/L,氯87 mmol/L,钙2.63 mmol/L,谷丙转氨酶35 U/L,谷草转氨酶48 U/L,三酰甘油1.77 mmol/L、总胆固醇5.54 mmol/L、血清肌酐53 μmol/L、eGFR 175.6 mL/(min·1.73m2)、葡萄糖4.1 mmol/L。肾素-血管紧张素-醛固酮系统(RAAS)检测结果见表1。

表1 肾素-血管紧张素-醛固酮水平
动脉血气分析 pH 7.47、氧分压91 mmHg、二氧化碳分压43 mmHg、实际碳酸氢根31 mmol/L、碱剩余6.8 mmol/L。
诊断分析患者父母是非近亲婚配,血钾均正常,该患者青少年男性,慢性起病,间断乏力三年,未正规诊治,此次起病前1周有腹泻病史,乏力症状较前加重,主要表现为低钾血症、代谢性碱中毒、低镁血症、低氯血症、低尿钙,血压正常,24h尿钾排出明显增多,故考虑为肾性失钾,结合患者血压正常、血气呈代谢性碱中毒、RAAS检测结果(表1),排除原发性醛固酮增多症(高血压、低肾素、高醛固酮水平),异位库欣综合征、肾上腺皮质癌、Liddle综合征(低肾素、低醛固酮水平),考虑为Batter综合征或Gitelman综合征,再根据患者24h尿钙偏低,有低镁血症,而Batter综合征多表现为高尿钙、血镁正常,故考虑Gitelman综合征可能性大。
初步诊断低钾血症,肾性失钾。
病情变化及诊疗经过患者入院后予以静脉及口服补钾治疗,共补氯化钾4 g/d,患者乏力症状逐渐好转,正常进食,1d后复查血钾2.9 mmol/L,继续补钾治疗2d后复查血钾2.4 mmol/L,予以加大每日补钾量,氯化钾6 g/d,2d后复查血钾2.7 mmol/L,患者乏力症状缓解。在正常进食,并予以补钾治疗的情况下,患者血钾仍然低于正常值,24h尿钾水平高于正常,进一步表明该患者的低血钾是肾性失钾所致,Batter综合征或Gitelman综合征可能性大,因而我们对该患者进行了基因检测。
基因检测口腔拭子取患者及其父母口腔黏膜上皮细胞行基因检测(Novocardio公司),共筛查了15种单基因高血压/血钾异常疾病(嗜铬细胞瘤综合征、家族性醛固酮增多症、Liddle综合征、Cushing综合征、多发性内分泌腺瘤病、家族性糖皮质激素抵抗征、Bartter综合征、Gordon综合征、Gitelman综合征等),包含41个相关基因,结果显示患者SLC12A3基因(Gitelman综合征相关致病基因)上存在1个杂合错义突变位点,该突变位点为第1 198位碱基G突变为T(c.1198G>T),对应编码的氨基酸由天冬氨酸变为酪氨酸(p.Asp400Tyr),转录本为第10外显子。患者父亲也检出杂合变异,先证者母亲为野生纯合,不携带该变异。因而推测患者的杂合变异位点来源于其父亲,不是新发变异(图1)。此外,在此次检测所包含的41个基因中,还发现一个与多发性内分泌腺瘤病2型相关的1个杂合错义突变位点,该突变位点为第1 441位碱基C突变为G(c.1441C>G),对应编码的氨基酸由亮氨酸变为缬氨酸(p.Leu481Val),该变异根据目前的研究表明与疾病并无明确相关性。
查询ClinVar等公共数据库显示,c.1198G>T 变异会导致p.Asp400Tyr 错义突异。该变异在ExAC数据库东亚人群、gnomAD数据库东亚人群中频率为 0,千人项目中国人群、本地数据库均未收录(PM2)。但根据SIFT、Polyphen2_HVAR、Polyphen2_HDIV、M-CAP 软件预测,该变异对基因或基因产物有害(PP3)。
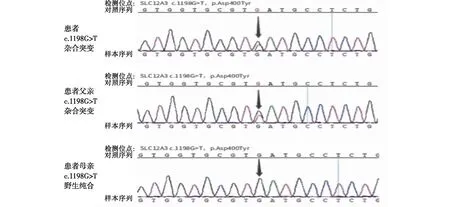
图1 Gitelman综合征患者及其父亲、母亲基因测序图
最后诊断Gitelman综合征。
后续治疗及随访出院后予以门冬氨酸钾镁片2片口服,3次/d,氯化钾缓释片1g 口服3次/d治疗1月,每周复查血钾,检测血钾波动在2.1~2.9 mmol/L,血镁0.59~0.62 mmol/L,患者无乏力、软瘫、抽搐等不适。1月后结合基因检测报告的结果,明确诊断为Gitelman综合征,遂加用螺内酯20 mg 3次/d,2周后复查血钾3.2 mml/L,血镁0.61 mmol/L。
讨 论
本文报道了1例临床诊断为Gitelman综合征的青少年,SLC12A3基因上存在1个突变位点,该突变位点为第1 198位碱基G突变为T(c.1198G>T),对应编码的氨基酸由天冬氨酸变为酪氨酸(p.Asp400Tyr),通过检索中国知网、Pubmed、Embase等数据库,发现该突变位点既往尚未见有关报道。
Gitelman综合征是一种常染色体隐性遗传性肾小管疾病,该病于1996年因Gitelman等报道了3例家族性低血钾、低血镁、低尿钙及代谢性碱中毒而得名[1]。Gitelman综合征的发病率至今尚未明确,国外有文献报道欧洲人群中发病率约1/40 000[2],亚洲人群中发病率可能高于该水平,根据日本的一项研究检提示人群中Gitelman杂合突变携带率达3.21%,推测10 000个受试者中有10.3个Gitelman综合征者,远远超过预期[3]。Lin等[4]通过对20名低钾血症的中国人群研究发现SLC12A3基因的杂合突变率达3%,Gitelman综合征的准确发病率仍需更大规模的研究才能确定。该病的发病基础是SLC12A3致病基因突变,导致其编码的肾远曲小管上皮细胞上的噻嗪类敏感性Na+/Cl-共转运体( NCCT) 功能失活,肾远曲小管重吸收氯化钠减少[5],K+和Mg+也在皮质集合管处大量丢失致使尿Mg+排出增多[6],血镁降低肾性失盐和血容量的减少可激活RAAS,进而导致低血钾及代谢性碱中毒[7]。由于NCCT异常可使细胞内Cl-的超级化作用减弱,Ca2+回吸收增加,尿钙减少。故临床以低钾血症碱中毒为主要表现,高肾素-血管紧张素-醛固酮水平,也有部分患者醛固酮水平也可表现为正常,肾脏活检病理检查可见肾小球旁器细胞增生[8]。多数患者在血钾稍低于正常值时并无明显不适症状,起病较为隐匿,因而很多患者可能并未得到及时诊断和治疗,由于技术及经济原因,基因诊断更是缺乏。本文总结了我科收治的1例青少年Gitelman综合征患者的临床资料及基因检测结果,并参阅相关文献分析了本病临床表型及可能存在的SLC12A3新突变位点。
本例患者间断乏力三年,平素无明显症状,此次起病前有腹泻病史,推测可能是腹泻使得在原本低血钾基础上,血钾经胃肠道丢失进一步减低至1.8 mmol/L,进而出现了四肢乏力,软瘫,实验室检查提示低血钾、低血镁、低氯性碱中毒,低尿钙(正常参考值2.5~7.5 mmol/24h),尿钾排泄增多(血钾<3.0 mmol/L时,24h尿钾>25 mmol),血压正常,RAAS系统检查提示肾素、血管紧张素Ⅱ明显升高,醛固酮正常。查阅相关文献发现Gitelman综合征患者也可表现为醛固酮不升高,这是由于远端肾小管对氯化钠重吸收减少,导致肾性失水失盐,低血容量,故导致RAAS激活,肾素、血管紧张素Ⅱ及醛固酮水平增加。然而,醛固酮也可以正常,这可能是低血钾对肾上腺分泌醛固酮有直接抑制作用,当抑制作用超过激活作用时也可表现为醛固酮正常[9]。该患者根据上述临床表现,临床考虑诊断为Gitelman综合征,经基因检测证实了诊断。
目前已发现400余个SLC12A3基因突变位点与Gitelman综合征相关,包括错义突变、剪切突变、无义突变、读码框位移突变等,其中以错义突变为主,这些突变分布在SLC12A3整个基因中,其中第1外显子和第10外显子的突变较为常见[10],该患者也为第10外显子突变,但是迄今为止并未发现任何突变热点,中国人群Gitelman综合征的高频突变位点为 T 60 M。一项最大样本的中国Gitelman综合征患者的基因型、表型和随访研究,发现 p.T60M、p.D486N、p.R913Q、c.965-1 c.977 delGCGGACATTTTTGinsACCGAAAATTTT以及 c.2877 2878delAG是中国人群中最常见的突变位点,该患者的突变位点并非常见位点[11]
SLC12A3基因突变导致蛋白功能减弱或丢失的潜在机制较为复杂,在目前发现的所有SLC12A3基因突变中,仅对其中的40种进行了功能评估[12-13],既往有研究表明在中国人群中,2 个等位基因均发现突变的患者占 83.6% ,只发现1个突变的患者占比也高达 16.4%[11]。该患者及其父母的基因检测结果提示患者及其父亲的SLC12A3基因上存在1个相同的突变位点(c.1198G>T),该患者出现了相应的表型,但是其父亲没有。曾有研究者指出,携带相同突变的患者临床表现不同,目前机制上不清楚,推测可能与以下因素相关[14]:(1)多数遗传性疾病与相应的基因编码区异常导致氨基酸改变相关,但是基因调节区如启动子、增强子、内含子又或者是5’3’端非编码区也可以干扰蛋白质的转录,推测这可能是该患者与其父亲出现不同表型的原因。(2)一种表型的出现往往是多个基因相互作用的结果,该患者也可能合并存在其他基因突变,而其父亲没有,与SLC12A3基因共同作用导致基因表达产物的异常,进而导致疾病的发生。(3)NCCT的表达和功能可能受饮食、环境和后天性修饰的影响,患者与其父亲这些方面存在差异,导致两人虽具有相同的基因突变,但其父亲尚未发病。
即往研究表明Gitelman综合征患者经积极补钾治疗后,低血钾和低血镁仍然难以纠正至完全正常,增加安体舒通的治疗均可进一步提高血钾和血镁水平,对该患者随访1月结果与之相一致,进一步验证了该患者Gitelman综合征的诊断。根据2017年1月发表在《Kidney International》上的KDIGO专家共识,认为Gitelman综合征合理的补钾目标值是3.0 mmol/L,补镁目标值是0.6 mmol/L。长期的低血钾可导致远曲小管以外的肾小管损害,该患者入院后肾小管功能检查也提示小管功能受损,长期的肾小管间质病变和低灌注可累及肾小球,出现蛋白尿及肾小球硬化,可进展至慢性肾功能不全;另外,肾素-醛固酮系统的激活也可能加重肾脏损害和纤维化,因而需要早期发现,明确诊断,早期治疗以改善预后。本例患者低钾血症的治疗,使患者得到及时正确的治疗,并发现了SLC12A3基因突变一个新的突变位点,丰富了Gitelman综合征基因突变谱,但该报道仅包括1例患者,关于该突变位点的致病性尚需更大样本量的进一步研究。
猜你喜欢
世界睡眠医学杂志(2021年4期)2021-07-03
种子(2021年3期)2021-04-12
透析与人工器官(2020年1期)2020-11-16
中华养生保健(2020年2期)2020-11-16
世界科学技术-中医药现代化(2020年2期)2020-07-25
牡丹江医学院学报(2020年2期)2020-07-01
中国循环杂志(2020年4期)2020-04-27
养生大世界(2019年12期)2019-12-11
中国生育健康杂志(2018年6期)2018-11-13
家庭科学·新健康(2018年8期)2018-10-30
