
木犀草素磷脂复合物的制备、表征及体内药动学行为
2019-07-23 11:42李阳杰
中成药 2019年6期
李阳杰, 周 敬
(郑州工业应用技术学院,药学与化学工程学院,河南 郑州451150)
木犀草素是黄酮类化合物,主要存在于白毛夏枯草、金银花、野菊花等植物中,具有抗炎、抗氧化、抗肿瘤、保肝等多种药理作用[1-2],临床适用于慢性支气管炎、支气管哮喘患者镇咳、祛痰[3],但其水溶性和脂溶性均较低[4],导致药物吸收存在较大问题[5]。磷脂复合物是中药活性成分和磷脂通过氢键等分子间作用力结合形成的不同于原型药物的一种复合物,可使中药活性成分由结晶状态转变为无定型状态,从而促进吸收,增加口服吸收生物利用度[6-13],但其在水相中的稳定性存在一定问题,故选择合适给药形式对其生物利用度有重大影响[9-10]。孙宇薇等[4]对木犀草素磷脂复合物进行了初步研究,但复合率很低,仅约77%;本实验重新对处方工艺进行研究,采用正交试验优化木犀草素磷脂复合物制备工艺,并考察该制剂基本性质和体内药动学行为,以期为其药效学研究提供参考。
1 材料
1.1 仪器 Agilent 1260 型高效液相色谱仪,配置DAD 检测器(美国Agilent 公司);DF-101S 型磁力搅拌器(巩义市予华仪器有限责任公司);NAI-DCY-12Y 型圆形水浴氮吹仪(上海那艾精密仪器有限公司);THZ-92A 型恒温振荡器(上海翠柳实验仪器有限公司);XW-80A 型漩涡混合器(上海医科大学仪器厂);D8 型X 射线粉末衍射仪(瑞士Bruker 公司)。
1.2 材料 木犀草素对照品(批号111520-201605,含有量99.6%,中国食品药品检定研究院);香叶木素对照品(批号P0587,含有量99.4%,上海源叶生物科技有限公司);卵磷脂(批号PC-98T,辅必成上海医药科技有限公司)。四氢呋喃为色谱纯(德国Merck 公司);司盘80(批号S-20161202,临沂优尼特生物科技有限公司);正辛醇、盐酸、高氯酸等(国药集团化学试剂有限公司)。
1.3 动物 SD 大鼠,雌雄兼具,体质量(300±20) g,购自上海斯莱克实验动物有限公司,动物生产许可证号SCXK(沪)2012-0002。
2 方法与结果
2.1 木犀草素含有量测定
2.1.1 色谱条件 Agilent ZORBAX SB C18色谱柱(250 mm×4.6 mm,5 μm);流动相乙腈-0.1%磷酸二氢钾(7 ∶3);检测波长348 nm;体积流量1.0 mL/min;柱温30 ℃;进样量20 μL。
2.1.2 方法学考察 精密称取木犀草素对照品25.0 mg,置于100.0 mL 量瓶中,加入约70 mL 乙腈超声溶解,静置至室温后定容,即得250.0 μg/mL 贮备液,精密量取适量,进一步稀释成5.0、10.0、25.0、50.0、125.0 μg/mL,在“2.1.1” 项色谱条件下进样测定。以峰面积(Y) 对溶液质量浓度(X) 进行线性回归,得方程为Y=0.030 6X-0.002 7(r=0.999 8),在5.0~125.0 μg/mL 范围内呈良好的线性关系。
然后,分别取高(125.0 μg/mL)、中(50.0 μg/mL)、低(5.0 μg/mL) 质量浓度对照品溶液,在“2.1.1” 项色谱条件下进样测定。结果,日内精密度RSD 为0.51%、0.32%、0.66%, 日间精密度RSD 为0.67%、 0.52%、0.93%,3 种质量浓度溶液的平均加样回收率分别为100.22%、99.64%、99.40%,RSD 分别为0.71%、0.84%、1.42%,表明该方法稳定可靠。
2.2 复合率测定 称取适量木犀草素与大豆卵磷脂在一定条件下进行反应,即得磷脂复合物,加入5 mL 二氯甲烷,振荡溶解,0.22 μm 微孔滤膜过滤除去未参加复合的木犀草素,收集滤液,除去二氯甲烷,收集固体,并加入5 mL乙腈,称定质量后继续振荡溶解,补加乙腈至振荡前质量,在“2.1.1” 项色谱条件下进样测定,测定参加复合的木犀草素质量,计算复合率,公式为复合率=X1/X0×100%。其中,X1为磷脂复合物中木犀草素质量,X0是木犀草素总质量。
2.3 正交试验 采用溶剂挥发法制备磷脂复合物。称取适量木犀草素和大豆卵磷脂,加入四氢呋喃,在一定温度条件下恒温搅拌,减压旋蒸,除去有机溶剂,40 ℃真空干燥箱中过夜干燥,即得,于干燥器中保存备用。
前期预实验发现,磷脂复合物复合率主要受搅拌时间(A)、搅拌温度(B)、木犀草素质量浓度(C)、木犀草素与大豆卵磷脂比例(D) 影响,故以四者为影响因素设计正交试验,并以复合率为评价指标。因素水平见表1,结果见表2,方差分析见表3。
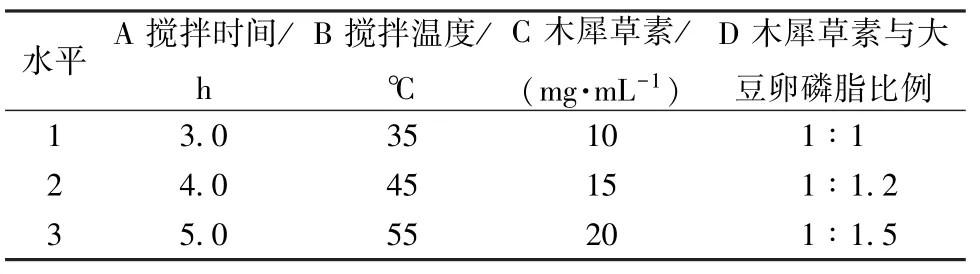
表1 因素水平
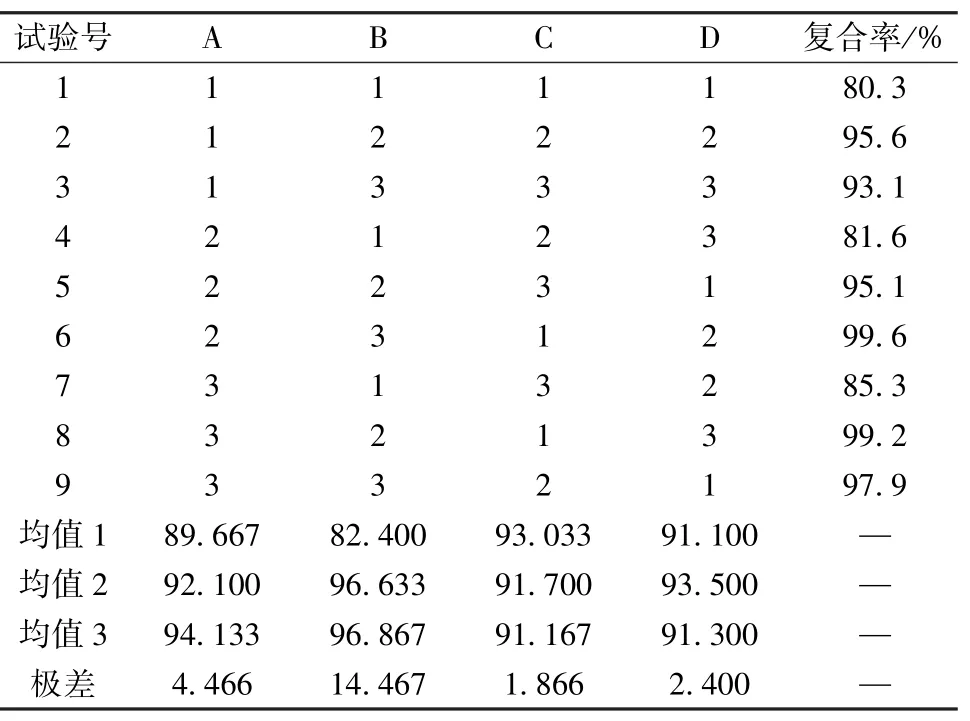
表2 试验设计及结果
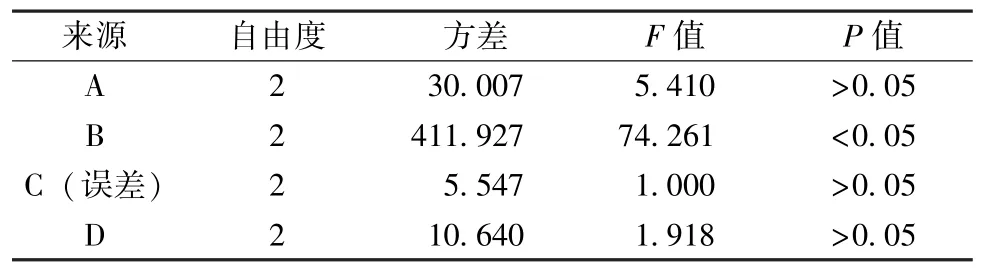
表3 方差分析
由表可知,因素B 对复合率有显著影响(P<0.05),而其他3 种因素无显著影响 (P >0.05);最优工艺为A3B3C2D2,即按1 ∶1.2 比例称取木犀草素、大豆卵磷脂,加入适量四氢呋喃使前者质量浓度为15 mg/mL,在55 ℃下恒温搅拌5 h,减压旋蒸除去有机溶剂,40 ℃真空干燥箱中过夜干燥,复合率均接近100%。
2.4 X 射线衍射(XRD) 分析 检测条件为Cu-Kα 靶,电流40 mA,扫描范围3°~45°,扫描速度8°/min,结果见图1。由图可知,木犀草素XRD 图谱有许多晶型峰,以结晶状态存在,可能是因为木犀草素、大豆卵磷脂极性端发生了作用,导致磷脂复合物中两者特征晶型峰均消失,最终呈现无定型状态,也证明磷脂复合物制备成功。
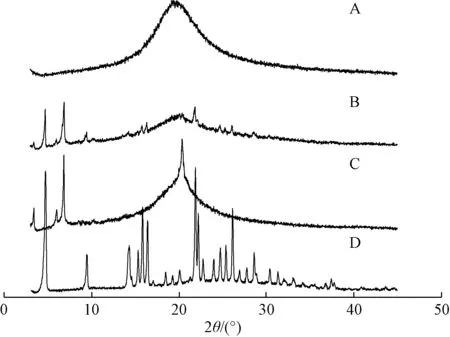
图1 样品XRD 图
2.5 溶解性测定 将过量木犀草素、物理混合物、磷脂复合物置于三角瓶中,加入蒸馏水(或正辛醇),室温下磁力搅拌24 h,6 000 r/min 离心15 min,小心吸取上清液,过滤,在“2.1.1” 项色谱条件下进样测定,计算表观溶解度,结果见表4。由表可知,磷脂复合物在水中的表观溶解度较木犀草素显著提高(P<0.01),同时在正辛醇中也显著高于在水中(P<0.01)。
表4 样品表观溶解度测定结果

表4 样品表观溶解度测定结果
注:与同一溶剂中木犀草素比较,∗∗P<0.01;与水中磷脂复合物比较,##P<0.01
样品 水/(μg·mL-1) 正辛醇/(μg·mL-1)木犀草素 53.83±0.42 511.51±3.14物磷理脂混复合合物物 1 71 03..19 74± ±01..52 57 ∗∗ 4 56 97 01..85 85± ±61.8 4.3 46∗∗##
2.6 体内药动学行为
2.6.1 灌胃供试液制备 取木犀草素适量,置于含0.5%CMC-Na 的蒸馏水中,超声分散,配制成15 mg/mL 混悬液,同法配制15 mg/mL(以木犀草素计) 磷脂复合物混悬液;取磷脂复合物适量,加入0.2 mL 司盘80,轻微研磨后置于中链甘油三酯中超声溶解,配制成15 mg/mL(以木犀草素计) 澄清状态的油制剂。
2.6.2 给药及取血[11]18 只大鼠于实验室环境中适应1 d,禁食不禁水,按30 mg/kg 剂量分别灌胃给予木犀草素混悬液、磷脂复合物混悬液、磷脂复合物油制剂,于0.15、0.5、0.75、1、1.5、2、4、6、8、12 h 眼眶取血各约0.3 mL,置于肝素化离心管中,4 000 r/min 离心4 min,分取上层血浆,-20 ℃冰箱中冷冻保存。
2.6.3 样品处理 精密吸取大鼠血浆样品100 μL、内标溶液20 μL,置于离心管中,加入3 mol/L HCl 溶液100 μL,涡旋混匀后在75 ℃水浴中放置2.5 h,加入5%HClO4溶液150 μL,3.0 mL 乙酸乙酯提取,涡旋15 min,10 000 r/min离心12 min,转移上层有机相于另一空白离心管中,再加入1.0 mL 乙酸乙酯同法提取,合并提取液,氮气吹干,100 μL 流动相复溶。
2.6.4 内标溶液制备 精密称取香叶木素对照品25.0 mg,置于100 mL 量瓶中, 加入乙腈超声溶解定容, 得0.25 mg/mL贮备液,精密量取2.0 mL 于50 mL 量瓶中,混匀后定容,质量浓度为10 μg/mL,继续精密量取1.0 mL于10 mL 量瓶中,混匀后定容,即得(1.0 μg/mL)。
2.6.5 方法学考察 取“2.1.2” 项下50.0 μg/mL 对照品溶液,逐步稀释成25.0、12.5、5.0、1.25、0.25 μg/mL,各取20 μL,加入180 μL 大鼠空白血浆,按“2.6.3” 项下方法处理,在“2.1.1” 项色谱条件下进样测定。以木犀草素质量浓度为横坐标(X),木犀草素、内标峰面积比例为纵坐标 (Y) 进行回归, 得方程为Y=1.162 2X+11.652 0(r=0.991 3),在25.0~2 500 ng/mL 范围间呈良好的线性关系。空白血浆、 对照品溶液、 供试品溶液HPLC 色谱图见图2,可知血浆内源性物质不干扰木犀草素和内标色谱峰,方法专属性良好。
取含25、150、 2 500 ng/mL 木 犀 草 素 的 血 浆, 在“2.1.1” 项色谱条件下进样测定。结果,3 种质量浓度日内精密度RSD(n=3) 分别为9.91%、5.32%、6.08%,日间精密度RSD(n=3) 分别为9.17%、8.69%、7.34%;将实际测定的质量浓度与配制的质量浓度比较,测得平均加样回收率在88.16%~93.09%之间;以信噪比S/N≥10 为定量限,S/N≥3 为检测限,测得两者分别为2.5、1.25 ng/mL。
2.6.6 考察方法 绘制血药浓度-时间曲线,计算主要药动学参数,结果分别见图3、表5。由此可知,磷脂复合物混悬液Cmax、AUC0~t、AUC0~∞与木犀草素混悬液比较均有所提高,但不明显(P>0.05);磷脂复合物油制剂tmax、Cmax、AUC0~t、AUC0~∞与磷脂复合物、木犀草素混悬液比较均显著升高 (P <0.05,P <0.01),生物利用度提高2.09 倍。
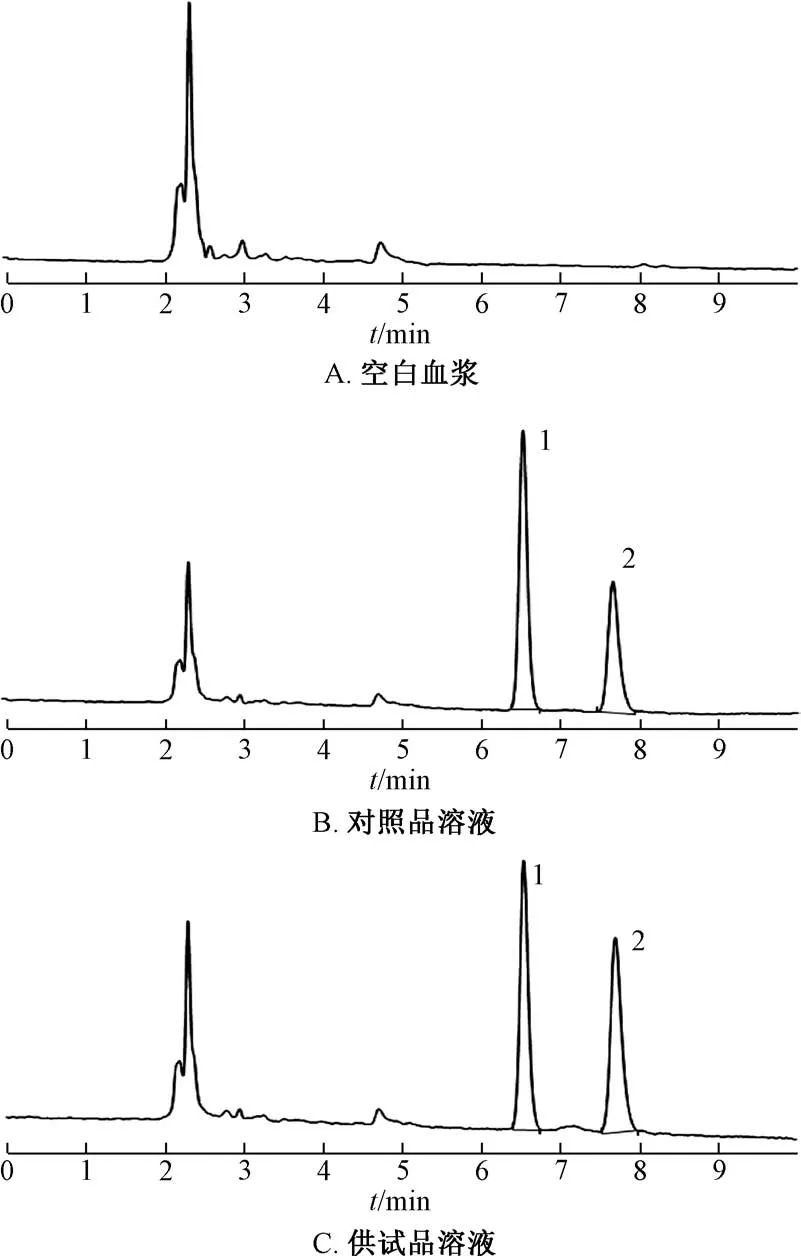
图2 木犀草素HPLC 色谱图
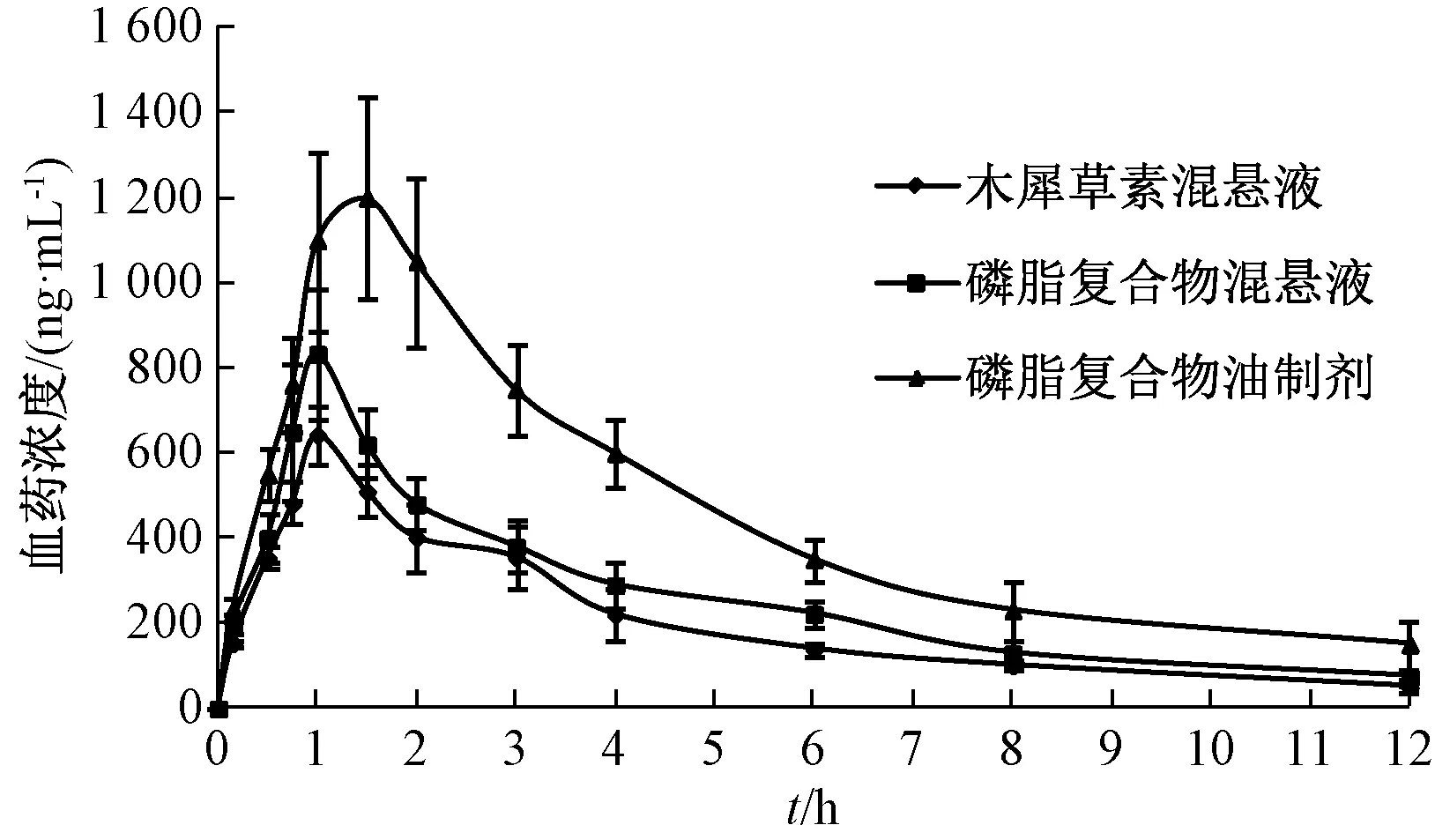
图3 样品血药浓度-时间曲线
表5 样品主要药动学参数
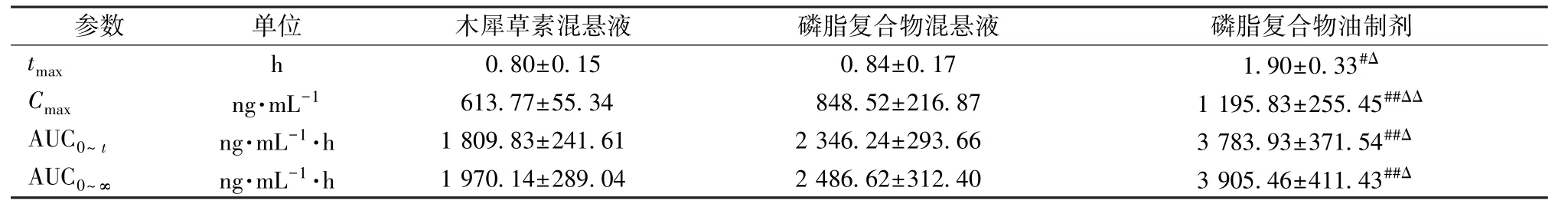
表5 样品主要药动学参数
注:与木犀草素混悬液比较,#P<0.05,##P<0.01;与磷脂复合物混悬液比较,ΔP<0.05,ΔΔP<0.01
参数 单位 木犀草素混悬液 磷脂复合物混悬液 磷脂复合物油制剂tCmmaxa x ng·m h L-1 61 03..87 07± ±05.5 1.53 4 84 08..85 42± ±02.1 16 7. 87 1 19 15..98 03± ±02.5 35 3.4#Δ 5##ΔΔ AUC0~t ng·mL-1·h 1 809.83±241.61 2 346.24±293.66 3 783.93±371.54##Δ AUC0~∞ ng·mL-1·h 1 970.14±289.04 2 486.62±312.40 3 905.46±411.43##Δ
3 讨论
本实验将制备溶剂四氢呋喃替换了无水乙醇,同时提高制备温度,此时木犀草素磷脂复合物复合率接近100%,高于无水乙醇中,其原因可能是乙醇分子中有可作为氢键给体的-OH 键,在一定程度上干扰了药物与磷脂形成复合物,导致复合率相对较低。木犀草素在血浆中容易与血清白蛋白形成一种结合物,为了准确测定血药浓度,必须使该成分从结合态游离出来,故在进行血浆样品处理时,依次采用盐酸、高氯酸对蛋白进行水解沉淀,取得了良好的结果。
另外,木犀草素磷脂复合物混悬液Cmax、 AUC0~t、AUC0~∞与木犀草素混悬液比较无明显差异,这可能是由于木犀草素与磷脂之间的作用力太弱,导致磷脂复合物以混悬液形式给药时由于水相干扰,使药物与磷脂发生了解离;以油制剂形式给药时,可避免水相对其干扰,为顺利透膜吸收奠定了基础。
猜你喜欢
今日畜牧兽医(2022年10期)2022-12-23
中国药学药品知识仓库(2022年5期)2022-04-11
北京联合大学学报(2022年1期)2022-02-12
烟台大学学报(自然科学与工程版)(2021年1期)2021-03-19
分析化学(2018年12期)2018-01-22
西江月(2017年4期)2017-11-22
中国中药杂志(2016年22期)2017-02-13
红豆(2017年1期)2017-01-14
小雪花·成长指南(2016年10期)2016-11-01
中国民族民间医药·下半月(2014年5期)2014-12-02
