
米托蒽醌的合成及质量评价Δ
2019-07-27 02:45王明明蔡锦源刘京顾浦中侍慧慧张艳军南京医科大学康达学院江苏连云港000广西科技大学鹿山学院广西柳州54566
中国药房 2019年13期
王明明,蔡锦源,刘京,顾浦中,侍慧慧,张艳军(.南京医科大学康达学院,江苏连云港000;.广西科技大学鹿山学院,广西柳州 54566)
血癌(白血病,Leukemia)是一种恶性肿瘤,是由克隆性白血病细胞大量增殖分化,弥散侵入人体正常组织器官而引发造血功能障碍和器质性病变的严重危害人类健康的一种疾病[1-2]。临床上表现为发热并引发细菌感染、贫血以及全身性多脏器的出血、淋巴结的肿大以及肝脾肿大等特征,目前临床治疗主要采用化疗和骨髓移植的方案,且以化疗为主[3-5]。
米托蒽醌(Mitoxantrone)又名丝裂蒽醌,是一种人工合成的蒽醌类化疗药物,具有抗肿瘤作用强、心脏毒性低的优点,用于治疗晚期乳腺癌、慢性淋巴细胞白血病(Chronic lymphocytic leukemia,CLL)和成人急性非淋巴细胞白血病等[6-7]。米托蒽醌进入人体后聚集在细胞核内,抑制DNA拓扑异构酶Ⅱ,通过Cheng氏N-O-O三角活性药效团[8-10]嵌入DNA的双链结构,配对DNA碱基对,使碱基对之间的距离由0.34 nm增至0.68 nm,引起DNA的裂解[11-13],阻断DNA与RNA的合成。米托蒽醌的N-O-O三角活性药效团及其嵌入DNA的示意图见图1。
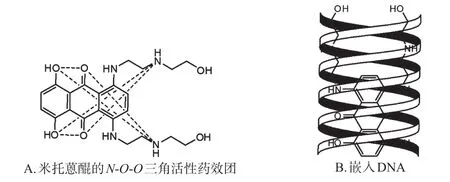
图1 米托蒽醌的N-O-O三角活性药效团及其嵌入DNA的示意图Fig 1 Triangular structure of N-O-O activity of mitoxantrone and DNAembedding diagram
在已报道的米托蒽醌的合成方法中郑卫等[14]研究了米托蒽醌类似物的合成,以4-甲基邻苯二甲酸为原料,经六步反应后得到化合物1,4-二羟基-6-氨乙基-9,10-蒽二酮,六步综合收率为12.6%,合成步骤长、副产物多、收率低。本试验采用1,4,5,8-四羟基蒽醌为原料优化米托蒽醌的合成工艺,以期缩减合成步骤、提高收率、降低生产成本,并检验合成出的多批次样品的理化特性,采用高效液相色谱法(HPLC)建立含量测定的方法,为米托蒽醌的合成和质量控制提供科学依据。
1 材料
1.1 仪器
1220型HPLC仪(美国Agilent公司);IRAffinity-1S型傅里叶变换红外光谱仪和AP-125WD型万分之一分析天平(日本Shimadzu公司);ZF-20D型紫外可见全波段分析仪(上海远怀化工科技有限公司);WRR型熔点测定仪(上海精科实业有限公司);Smart-Q型去离子纯水机(美国The-Lab公司);GZX-9070MBE型电热鼓风干燥箱(上海博迅实业有限公司);RE-52AA型旋转蒸发器(上海亚荣生化仪器厂);SHB-Ⅲ型循环水式真空泵(巩义予华仪器有限责任公司);HH-4型数显恒温水浴锅(国华电器有限公司);KQ5200型超声清洗器(昆山市超声仪器有限公司);TGL-16C型高速离心机(上海安亭科学仪器厂)。
1.2 药品与试剂
盐酸米托蒽醌对照品(中国食品药品检定研究院,批号:101134-201001,纯度:98.9%);1,4,5,8-四羟基蒽醌(山东西亚化学股份有限公司,批号:M3290,纯度:96.0%);1,4-二氧六环、N-(2-羟乙基)乙二胺、正己烷、无水硫酸钙、无水甲醇、醋酸铵、乙腈(国药集团化学试剂有限公司);水为纯化水。
2 方法与结果
2.1 米托蒽醌的合成
2.1.1 米托蒽醌粗品的制备 称取1,4,5,8-四羟基蒽醌 5 g(0.018 mol),氩气保护下加入 1,4-二氧六环 32 mL,磁力搅拌至固体完全溶解,恒压滴定漏斗逐滴加入N-(2-羟乙基)乙二胺15 g(0.15 mol),反应液为糊状黏稠液体。持续搅拌,水浴50℃反应2 h,反应液为蓝褐色油状物,加入100 mL无水乙醇,调节水浴温度至55℃,通入干空气(无水硫酸钙作为干燥剂)缓慢氧化4 h,反应液由蓝褐色转换为亮蓝色。反应液放冷补足乙醇余量,将混合物转移至250 mL广口瓶中,室温条件下放置24 h,待瓶底有明显的固体沉降物,减压抽滤得蓝色结晶,即米托蒽醌粗品。米托蒽醌合成路线图见图2。
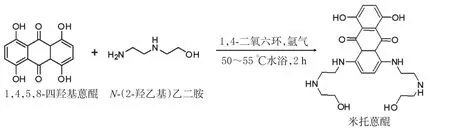
图2 米托蒽醌合成路线图Fig 2 Synthesis pathway of mitoxantrone
2.1.2 米托蒽醌粗品的精制 取米托蒽醌粗品结晶9.2 g,置于茄型瓶中,加入乙醇-正己烷(4∶1)混合液200 mL,加热至完全溶解,加入适量活性炭,热回流15 min,趁热抽滤,滤液置于冷水中过夜析晶,抽滤得米托蒽醌晶体,晶体用乙醇-正己烷(3∶1)冲洗3次,得深蓝色至蓝黑色米托蒽醌粉末状晶体2.8 g,产率为34.3%,显微镜观察,调节目镜倍率×10,物镜倍率为×40,米托蒽醌微观结构为分散状晶态颗粒聚集体,颗粒大小为μm级,米托蒽醌粉末的显微镜图见图3。分别制备米托蒽醌供试品4批,批号分别为171102、171106、171112、180313。
2.2 米托蒽醌的理化特性以及结构鉴定
2.2.1 熔点测定 取干燥米托蒽醌供试品(批号:180313)适量,研细成粉末,置于一端封口的毛细管中,熔点仪测定熔点范围为159.1~163.6℃,相对于米托蒽醌标准熔点170~174℃,盐酸米托蒽醌标准品203~205℃,熔程偏宽,熔点明显降低,表明药物结晶中可能包埋有降低熔点的杂质,难以通过重结晶方式除去。

图3 米托蒽醌粉末的显微镜图Fig 3 Microscopic morphology of mitoxantrone
2.2.2 pH测定 取各批次米托蒽醌供试品粉末0.05 g,加水25 mL振荡使完全溶解,制备成每1 mL含2 mg药物的溶液,中性滤纸滤过,搅拌均匀,室温条件下放置5 min,待溶液澄明稳定,作为pH测定液A。取测定液A加水稀释100倍,同法操作,作为pH测定液B。分别测定测定液A和测定液B的pH值,每批次样品各测3次。结果表明,米托蒽醌高浓度条件下(测定液A)偏碱性,稀释后水溶液(测定液B)近中性,合成的批次产品间pH差异小,室温下溶液短时期内理化特性稳定。不同批次米托蒽醌水溶液pH值的测定结果见表1。
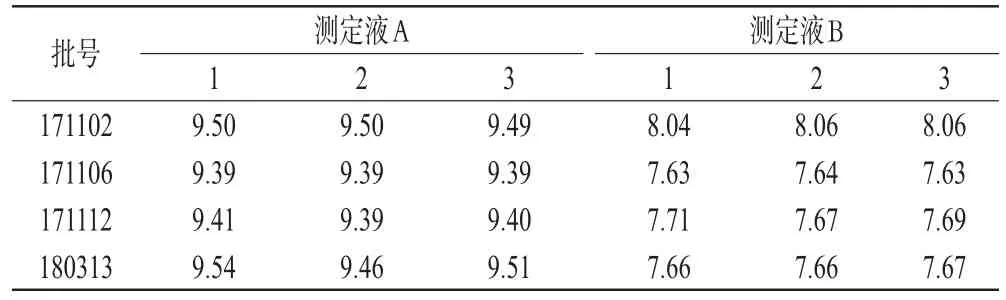
表1 不同批次米托蒽醌水溶液pH值的测定结果Tab 1 pH value of different batches of mitoxantrone solution
2.2.3 紫外-可见吸收光谱的检测 取各批次米托蒽醌供试品,加水溶解制成每1 mL中含0.2 mg的溶液,置于吸收池中,调节紫外-可见波段为200~800 nm,扫描速度为快速,采样间隔为0.5 nm进行扫描。不同批次米托蒽醌水溶液的紫外-可见光全波段扫描光谱图见图4。
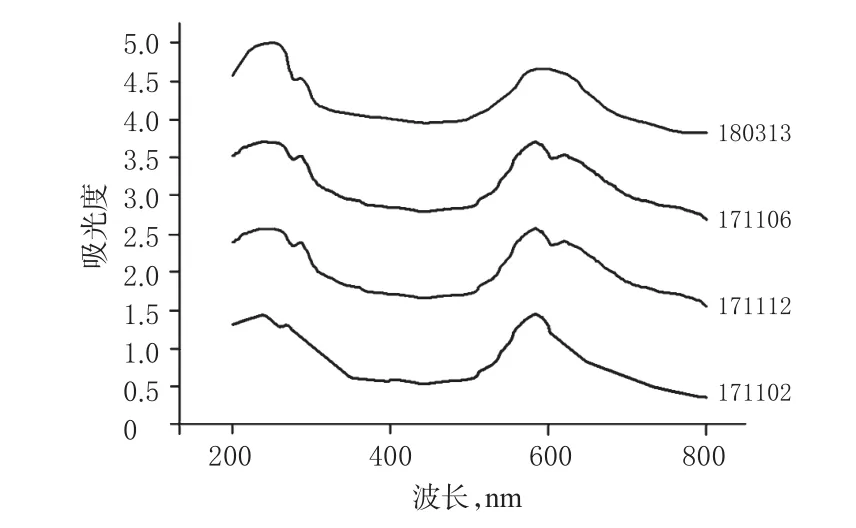
图4 不同批次米托蒽醌水溶液的紫外-可见光全波段扫描光谱图Fig 4 UV full-band screening spectrum of different batches of mitoxantrone solution
图4显示,4批次的米托蒽醌均在紫外波段235~245 nm范围内出现紫外最大吸收峰,268 nm波长处附近存在肩峰,在可见光波段590~600 nm波长范围内出现可见光最大吸收峰,部分样品在680 nm波长处出现可见光的等吸收点,提示4批次米托蒽醌的紫外-可见波段光谱吸收曲线基本保持一致。
2.2.4 红外结构特征的鉴定 取米托蒽醌供试品(批号:180313)1 mg,置于玛瑙研钵中,加入干燥的溴化钾粉末约200 mg,充分研磨,压片,使用傅里叶变换红外光谱仪检测红外光谱,米托蒽醌的红外光谱图见图5。

图5 米托蒽醌的红外光谱图Fig 5 IR spectrum of mitoxantrone
图5显示,米托蒽醌的红外特征与《药品红外光谱集》(2015年版)中米托蒽醌项下描述一致,其中3 317 cm-1处为饱和烃碳支链末端羟基和羟基的吸收峰,峰形尖锐,向低波数产生位移,游离脂肪族羟基与酚羟基有一定程度的缔合,但产品在固体状态下缔合力弱,故不会产生明显的宽峰;2 940 cm-1和2 859 cm-1处为仲胺基团的吸收峰,这是由于苯环上连接的仲胺基团受到苯环电子团的共轭作用,与芳杂环连接的近端氨基与远端氨基发生分离,产生尖锐的双峰;1 609、1 569、1 475、1 448.82 cm-1为骨架蒽醌环的特征吸收峰,本品研磨压片时所用的量低于常规压片用量,因为本品透光率差,常规浓度下吸收峰透光率低易掩蔽特征吸收。
2.2.5 干燥失重的考察 取各批次的干燥米托蒽醌供试品粉末约1.0 g,均匀分散,平铺于直径为5 cm的扁平称量瓶中,置于105℃干燥箱中干燥2 h至恒重,取出置于密封干燥器中放冷,精密称定,计算失水率[(干燥前质量-干燥后质量)/干燥前质量×100%]。不同批次米托蒽醌干燥失重结果见表2。

表2 不同批次米托蒽醌干燥失重结果Tab 2 Drying weightlessness of different batches of mitoxantrone
2.2.6 临界相对湿度(CRH)的测定 ①恒温恒湿条件:制备CH3COOK、MgCl2、K2CO3、NaBr、KCl、KNO3的饱和盐溶液各1份,分别置于密闭干燥器中,设定温度25℃。各盐溶液对应湿度关系见表3。②吸湿平衡时间的测定:精密称取干燥至恒重的米托蒽醌供试品粉末(批号:180313)约50 mg,均匀铺散于干燥至恒重的敞口具塞称量瓶中,再置于温度25℃、相对湿度75%的密闭环境中,每隔4 h取出称量瓶,精密称定。考察样品于不同时间点(0、6、9、21、28、36、52、61 h)的累积吸湿量,计算吸湿百分率[(吸湿后质量-吸湿前质量)/吸湿前质量×100%],以放置时间为横坐标,吸湿百分率为纵坐标,绘制吸湿平衡曲线,找出样品吸湿量处于平衡状态下所对应的时间折点为吸湿平衡时间。结果显示,米托蒽醌的吸湿平衡时间为48 h。米托蒽醌在温度25℃、相对湿度75%条件下放置不同时间的吸湿百分率见表4,对应的吸湿平衡曲线见图6。③CRH的测定:精密称取干燥至恒重的米托蒽醌供试品粉末(批号:180313)约50 mg,共计6份,同②法操作,分别置于①中6种饱和盐溶液密闭湿度环境中,室温25℃放置48 h,取出后精密称定。以环境湿度为横坐标,样品吸湿百分率为纵坐标,绘制CRH曲线,在CRH曲线的高低湿度处选取一点作切线,两切线交于一点,该交点的横坐标对应的环境湿度记作米托蒽醌的CRH值。温度25℃下米托蒽醌在不同盐溶液(湿度)下的吸湿百分率见表3,对应的CRH曲线见图7。
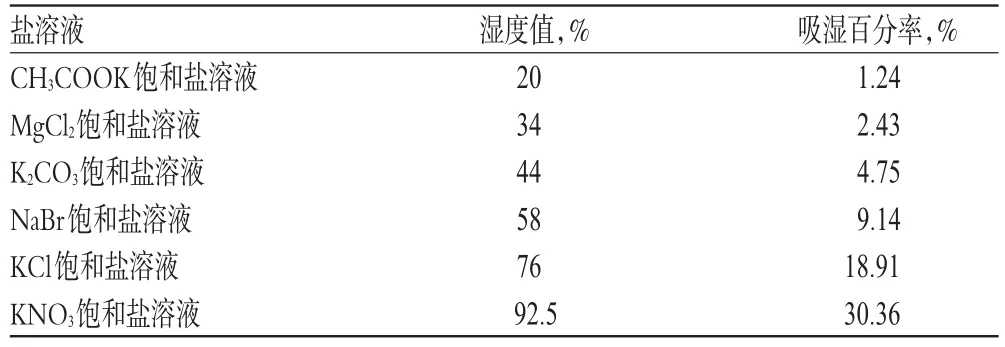
表3 温度25℃下米托蒽醌在不同盐溶液(湿度)下的吸湿百分率Tab 3 Hygroscopicity percentage of mitoxantrone in different salt solution(humidity)at 25 ℃

表4 米托蒽醌在温度25℃、相对湿度75%%条件下放置不同时间的吸湿百分率Tab 4 Hygroscopicity percentage of mitoxantrone placing for different time at 25℃with relative humidity of 75%%
图7显示,25℃下米托蒽醌的CRH为54.7%,吸湿百分率为2.4%,提示米托蒽醌在常温常湿条件下(温度25℃,相对湿度40%)贮存易吸收环境中的水份,具有引湿性,但不会潮解,长期存放应干燥后密封贮存。
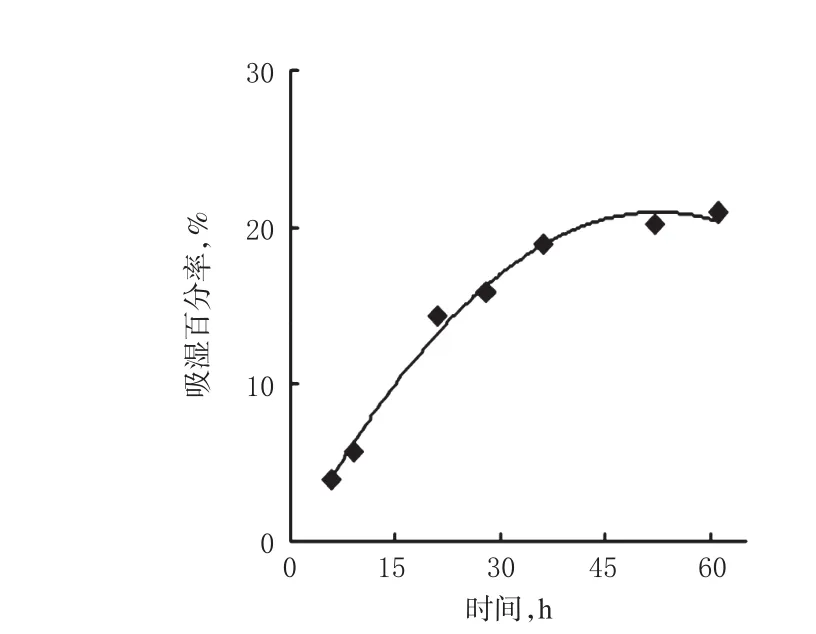
图6 米托蒽醌在温度25℃、相对湿度75%%条件下的吸湿平衡曲线Tab 6 Water absorption percentage of mitoxantrone placing for different time at 25℃with humidity of 75%%

图7 温度25℃下米托蒽醌在不同盐溶液(湿度)的CRH曲线Fig 7 Hygroscopic equilibrium curve of mitoxantrone in different salt solution(humidity)at 25 ℃
2.3 米托蒽醌的含量测定
2.3.1色谱条件色谱柱:Agilent BDS C18(250 mm×4.6 mm,5 μm);流动相:乙酸铵水溶液(0.2 mol/L,pH=4.4)-乙腈(1∶4,V/V);检测波长:242 nm;柱温:30℃;流速:0.5 mL/min;进样量:10 μL。
2.3.2 对照品溶液的制备 取米托蒽醌对照品25.43 mg,精密称定,置于100 mL量瓶中,水溶解稀释定容至刻度,摇匀,作为贮备液;精密量取1 mL贮备液,置于25 mL量瓶中,流动相溶解稀释至刻度,摇匀,0.22 μm有机滤膜过滤,作为对照品溶液。
2.3.3 供试品溶液的制备 取批号分别为180313、171102、171106、171112的米托蒽醌供试品各25 mg,精密称定,流动相稀释制成质量浓度均为10 μg/mL的溶液,作为供试品溶液。
2.3.4 系统适用性试验 用流动相制成米托蒽醌质量浓度为 0.01 mg/mL,1,4,5,8-四羟基蒽醌质量浓度为10.0 mg/mL的混合溶液作为系统适用性溶液。取系统适用性溶液、对照品溶液和供试品溶液(批号:180313),按“2.3.1”项下色谱条件进样测定,记录色谱图。结果显示,米托蒽醌出峰时间为10.23 min,理论板数以米托蒽醌峰计不低于6 000,对称因子大于0.75;1,4,5,8-四羟基蒽醌出峰时间为12.36 min,相邻峰分离度均大于2。色谱图见图8。

图8 高效液相色谱图Fig 8 HPLC chromatograms
2.3.5 线性关系考察 精密量取米托蒽醌贮备液0.5、0.8、1.0、1.2、1.5 mL,分别置于25 mL量瓶中,流动相溶解稀释定容至刻度,摇匀,0.22 μm有机滤膜过滤,作为50%、80%、100%、120%、150%系列标准曲线溶液。各精密量取系列标准曲线溶液10 μL进样,每份样品平行测定2次,以米托蒽醌峰面积(A)为纵坐标、质量浓度(c)为横坐标绘制标准曲线。得回归方程为A=6.555 0×103c-1.375 9(r=0.999 8),结果表明,米托蒽醌在质量浓度为5.086~15.258 μg/mL范围内保持良好的线性。
2.3.6 精密度试验 精密量取米托蒽醌对照品溶液10 μL,按照“2.3.1”项下色谱条件连续进样6次,记录峰面积。结果显示,峰面积的RSD为0.19%(n=6),表明仪器精密度符合要求。
2.3.7 准确度试验 精密量取已知含量的米托蒽醌水溶液(批号:180313)1 mL,置于100 mL量瓶中,水稀释溶解定容至刻度,精密量取上述溶液1 mL,共9份,分别置于100 mL量瓶中,精密称取米托蒽醌对照品约5.15、7.15、9.15 mg,各3份,分别置于上述9个100 mL量瓶中,水溶解混匀稀释至刻度,分别作为80%、100%、120%浓度水平的溶液,各精密量取1 mL置于10 mL量瓶中,流动相溶解稀释至刻度,进样10 μL,记录峰面积,代入回归方程计算含量,计算回收率。结果显示,米托蒽醌的平均回收率为99.85%,RSD%为1.69%(n=9),回收率试验结果见表5。

表5 回收率试验结果Tab 5 Results of recovery tests
2.3.8 耐用性试验 取供试品溶液(批号:180313),分别改变“2.3.1”项下色谱条件中的pH(水相pH改为4.4±0.2)、柱温[柱温改为(30±2)℃]、流动相比例[乙腈占流动相体积比改为(80±5)%范围内]等因素,考察本品测定结果在色谱条件改变下的耐受程度。结果表明,调节pH、柱温、流动相比例后米托蒽醌的出峰时间和峰形都稳定,表明该色谱条件耐用性良好。
2.3.9 含量测定 精密量取各批次米托蒽醌供试品溶液,按照“2.3.1”项下色谱条件进样测定,记录色谱图。主成分含量以对照品外标法计算,结果显示,批号分别为180313、171102、171106、171112的米托蒽醌供试品中米托蒽醌含量分别为83.2%、76.3%、73.9%、79.1%,平均值为78.1%。
3 讨论
本试验以1,4,5,8-四羟基蒽醌作为原料,隔绝空气(氩气保护)恒温的试验条件下制备出米托蒽醌粗品,其中氩气保护是必须的,研究发现通过氮气保护后制备的产品经过隔夜重结晶操作后产率只相对于氩气保护产率的50%~70%,这可能是由于反应混合液质地黏稠,流动性差,搅拌过程中易于掺杂气泡,而氩气的密度比重相对于氮气更大,可有效地排除体系中的空气,抑制羟基的酯化、氧化等副反应,促进C—N键的生成;同时反应温度也是至关重要的,50~55℃为最佳的反应温度,温度升高至60℃以上时,米托蒽醌产率降低,杂质增多,产品呈非晶体态的松散粉末;反应温度低于50℃时,原料反应不完全,反应液呈棕色。米托蒽醌粗品的制备需经过24 h的析晶操作促使晶体颗粒从乙醇溶液中析出,不宜采用冰水浴加速析出过程,室温下过长或过短时间的析晶过程易导致结晶的失败,最佳的结晶时长为12~24 h。
精制得到的米托蒽醌具有极强的水溶性,干燥后产品为粉末,易吸收空气中水分而使自身潮解,研究CRH能够更好地了解物料的特性,有效控制物料的贮存条件,防止药品在长期贮存过程中的潮解和霉变[15-18]。本研究结果显示,25℃下米托蒽醌的CRH为54.7%,提示该产品在潮湿环境中存放易吸潮和水化。相对于2015年版《中国药典》药物引湿性试验指导原则,本研究中CRH测定方法显著改善了测量样品的称样量,这是基于提高试验数值的准确性、减少系统误差的考虑,对于样品分散程度也作出了严格的规定,各批次试验中要求铺层的高度为2 mm,铺散层为直径为1 cm的圆饼状,这类处理有利于试验数值的一致性和平行性考察,同时该试验中的光谱及色谱学检测标准依据试验要求也作了合理的调整,有利于结果的观察和测定。本试验未考察有关物质指标,因考虑到合成中可能产生的杂质为米托蒽醌同分异构体,采用常规C18色谱柱分离检测,同分异构体检测峰之间会产生叠加和包埋现象,干扰结果测定,可考虑进一步采用手性分离色谱柱和质谱联用技术作为今后的研究内容。
米托蒽醌是一种人工合成的治疗淋巴瘤的特效药,采用全合成方法制备,合成路线长、副产物多、提纯困难,造成原料药价格昂贵[19-20]。本试验采用1,4,5,8-四羟基蒽醌作为合成原料,温和绿色的试验条件下制备的米托蒽醌产品经精制后产率高达34.3%,产品的理化特性经检验符合原料药用药需求,建立的HPLC分析方法适用于原料药含量的测定,4批米托蒽醌的平均含量为78.1%,其中批号180313的米托蒽醌供试品中米托蒽醌含量可达83.2%,产品纯净、杂质少。本试验中米托蒽醌的分析方法稳定性好、重复性好、分析快速、结果准确可靠,合成的产品纯度高、副产物低、制备工艺简单易行,可考虑将该方法用于进一步扩大化生产。
猜你喜欢
河南化工(2022年10期)2022-11-21
航天电子对抗(2022年4期)2022-10-24
河南农业·综合版(2022年2期)2022-03-18
化学分析计量(2021年5期)2021-05-24
西北药学杂志(2021年1期)2021-02-03
中华养生保健(2020年3期)2020-11-16
山西中医药大学学报(2020年2期)2020-06-06
中国科技纵横(2018年2期)2018-11-29
Asian Journal of Urology(2015年3期)2015-12-16
中国新技术新产品(2012年11期)2012-12-28
