
运用高通量测序技术研究一Waardenburg综合征家系
2019-08-13 07:48徐彬戴继任付勇
中华耳科学杂志 2019年4期
徐彬 戴继任 付勇
浙江大学医学院附属儿童医院(杭州310052)
耳聋是人类最常见的致残性疾病之一。在世界范围内,据统计新生儿先天性聋的发病率为1‰-3‰,50%患儿的耳聋与遗传因素有关[1]。根据是否合并有其他系统或器官疾病,临床上将遗传性耳聋分为综合征型聋(Syndromic Hearing Impairment,SHI)与非综合征型聋(Non-Syndromic Hearing Impairment,NSHI)两型。30%的遗传性耳聋患者为SHI,70%为 NSHI。
Waardenburg综合征(Waardenburg Syndrome,WS)又称听力-色素综合征,是一种常见的染色体显性遗传综合征遗传性聋,WS发病率无明显性别及种族差异,发病率占先天性耳聋患者的1%-3%,而人群发病率为1/42000[2]。WS具有高度的遗传异质性,主要临床表现为感音神经性听力损失及色素分布异常,依据其伴随症状的不同分为WSI、WSII、WSIII、WSIV四种类型[3],WSI临床表现为先天性感音神经性听力下降,色素异常及内眦异位,指数W>1.95[4];WSII与WSI的区别就是无内眦异位,W<1.95[5];WSIII除了WSI的临床症状外,还伴有上肢畸形[6];WSIV除了WSII的临床表现外,同时伴有消化道的异常,例如先天性巨结肠或胃肠道闭锁[7]。在Waardenburg综合征患儿中,虽有虹膜异色,但无明显的视力改变,且该疾病患儿的皮肤毛发色素分布异常也对其日常生活没有明显的影响,因此,此类患儿常常因为听力问题就诊,WS患儿中约20%-55%伴有重度或极重度感音神经性聋,占先天性聋儿的2%-5%[8]。
本研究对一Waardenburg综合征家系进行深入考察,采用高通量测序技术(Next-Generation Sequencing,NGS)以及Sanger测序方法检测该家系致病基因,并探讨其可能性分子遗传学机制。
1 对象与方法
1.1 研究对象收集来自浙江省温州市的1个Waardenburg综合征家系,命名为WZ-1家系,该家系共3代,现存家系成员21人,男女成员均有耳聋患者,系谱分析符合常染色体显性遗传规律,家系图(图1)。
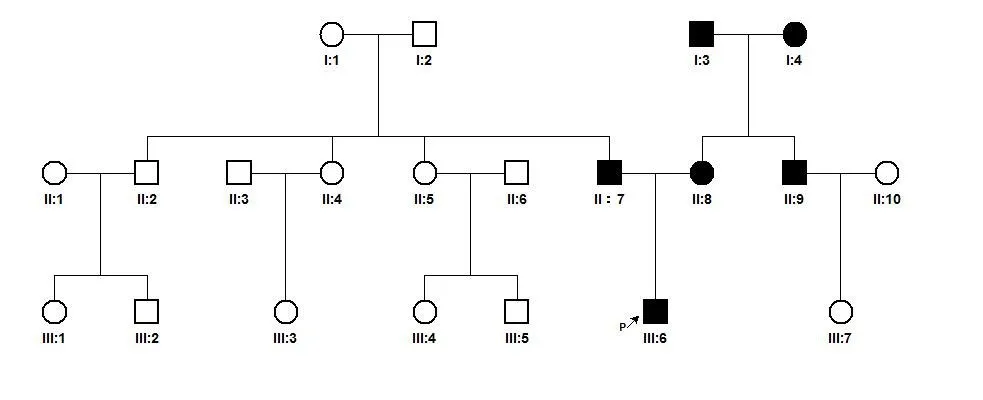
图1 WZ-1家系图谱●■耳聋患者P↗■先证者Fig.1 Pedigree of family WZ-1●■deaf patients P↗■ proband
1.2 研究方法
1.2.1 病史及听力学检测
对该家系人员采集信息及抽取血样前对家属进行宣教,告知其检查的目的、意义,对自愿接受检测者签署知情同意书。登记其一般情况,详细询问耳聋患者的发病年龄、是否存在诱因、耳聋的具体病变进展情况,有无相关伴随症状、既往有无外伤史、耳毒性药物服用史、出生时有无宫内缺氧及核黄疸等,在耳鼻喉科、眼科、皮肤科、消化内科、骨科进行系统的体格检查,根据耳聋患者内眦、外眦及瞳孔距离,计算W值。
先证者在我院行声导抗、OAE、ABR、ASSR及颞骨CT、头颅MRI等,排除颅脑占位性病变及智力障碍等。其家属在我院及温州医科大学附属第二医院完成相关体格检查及听力学检测。
听力诊断标准参考WHO1997年日内瓦会议推荐的听力损失分级标准。以纯音测听为基础,以较好耳0.5 、1.0、2.0、4.0kHz的听阈平均值为依据,将听力减退分为四个级别,即26~40dBHL为轻度听力损失;41~60dB HL为中度;61~80 dB HL为重度;≥81 dBHL为极重度。
1.2.2 基因检测
抽取先证者及其家属的外周血2-3ml,注入EDTA紫色抗凝管中,编号后送检。先证者采用高通量测序技术,检测先证者的159个基因的外显子区,6个线粒体基因,3个miRNA,再对先证者及其家属进行Sanger测序验证。
高通量测序流程如下:
①目标区域捕获测序
目标区域捕获测序的主要流程是利用捕获芯片将目标区域的DNA片段进行富集后再借助高通量二代测序平台进行测序。本研究采用目标序列捕获芯片对已知的耳聋候选基因进行捕获。捕获的具体过程如下:将基因组DNA随机打断成片段,与Illumina PE接头寡核苷酸混合物链接,对产物进行链接介导的聚合酶链式反应(LM-PCR)扩增纯化后得到DNA文库,并对其进行质量检测,将上述PCR产物与目标区域捕获芯片进行杂交以富集目标区域序列,借助Illumina Next 500测序平台对捕获的序列进行测序,并对原始数据进行初步处理,包括图像识别和样本区分。
②生物信息分析
将原始测序数据去除污染和接头序列,然后利用BWA软件将过滤后的序列比对到NCBI数据库人类基因组参考序列(hg19)上,利用GATK软件分析得出单核苷酸变异(SNV)和插入缺失突变(INDEL)的相关信息。然后通过ANNOVAR软件对所有的SNP和INDEL进行注释。筛选掉正常人数据库中频率小于0.05的突变位点,正常人数据库包括千人基因组计划、Exome Variant Server和EXAC。错义突变使用SIFT,PolyPhen-2 MutationTaster和GERP++软件进行致病性预测和保守性预测,剪切位点的改变用SPIDEX软件分析其致病性。
③PCR扩增和Sanger测序
经过分析筛选得到的获选变异位点利用PCR和Sanger测序验证。PCR引物对利用Primer 3.0在线软件设计。PCR产物经过Sanger测序并在ABI 3130 Genetic Analyzer(Applied Biosystems)上进行分析。再在家系成员中进行共分离验证。
2 结果
2.1 病史及辅助检查结果
该家系共3代,现存家系成员21人,系谱分析符合常染色体显性遗传特征。参与本研究的耳聋患者6人,有语前聋和语后聋。外貌特征先证者(III:6)左侧虹膜为亮蓝色(图3),内眦间距正常,W<1.95;其外公(I:3)头发、眉毛稀疏,色白。家系听力学检测结果提示:先证者出生时听力筛查未通过,1月后复筛未通过,6月时声导抗提示双耳A型曲线,OAE提示双耳均未通过,ABR及ASSR提示双耳极重度感音神经性聋,颞骨CT及头颅MRI提示未见明显异常;其余家属声导抗提示双耳A型曲线,OAE提示双耳均通过;妈妈(II:8)为先天性极重度感音神经性聋;爸爸(II:7)为极重度感音神经性聋,出生时听力正常,2岁时感冒用药后出现耳聋;舅舅(II:9)为轻度感音神经性聋,发病时间不详;外公为先天性极重度感音神经性聋;外婆(I:4)为中度感音神经性聋,5岁时发热后才出现耳聋,其余家属无耳聋病史(图2及表1)。
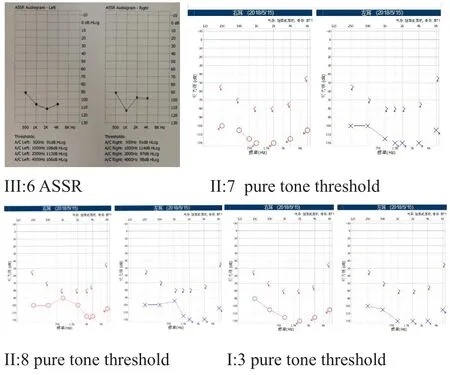
图2 WZ-1家系成员听力图Fig.2 audiogram of family WZ-1

表1 WZ-1家系患者基本资料Table 1 Basic data of family WZ-1

图3 先证者左侧虹膜为亮蓝色Fig.3 The proband's left iriswerebright blue
2.2 基因检测结果
先证者通过高通量测序发现MITF(c.397G>T)突变。后续经一代测序验证,其母亲、外公同样存在MITF(c.397G>T)突变。(图4)。
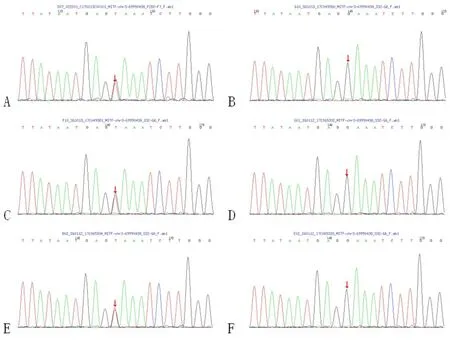
图4 Sanger测序验证结果A为先证者、C为先证者母亲、E为先证者外公发现MITF(c.397G>T)突变;B、D、F为先证者父亲、舅舅、外婆相同位点未发现突变。Fig.4 Results of Sanger sequencingA III:6 has M ITF(c.397G>T)mutation;B II:7 w ithout M ITF(c.397G>T)mutation;C II:8 has M ITF(c.397G>T)mutation;D II:9 w ithout M ITF(c.397G>T)mutation;E I:3 has M ITF(c.397G>T)mutation;FI:4 w ithout M ITF(c.397G>T)mutation.
3 讨论
WS的诊断主要标准包括:(1)先天性感音神经性耳聋;(2)虹膜色素异常:(3)头发低色素改变,表现为额白发;(4)内眦异位:W指数>1.95;(5)一级亲属患病,满足2个标准就能确诊WS[9]。该WS家系中先证者具有先天性极重度感音神经性聋、左侧亮蓝色虹膜符合两个WS主要诊断标准,患儿无内眦异位:W指数<1.95,排除WS I型,四肢无畸形活动正常,排除WSIII型,颞骨CT、头颅MRI提示未见异常,经相关检查排除先天性巨结肠和胃肠道闭锁,排除WS IV型,先证者确诊为WSII型。先证者外公先天性极重度感音神经性聋、额白发符合两个WS主要诊断标准,四肢无畸形活动正常,无先天性巨结肠和胃肠道闭锁,无内眦异位:W指数<1.95,同样为WSII型。先证者母亲先天性极重度感音神经性聋、一级亲属患病符合一个WS主要诊断标准,四肢活动无畸形活动正常,无先天性巨结肠和胃肠道闭锁,无内眦异位:W指数<1.95,亦为WSII型。
研究表明与WS相关的基因有6种:SOX10、PAX3、MITF、SNAI2、EDNRB和EDN3[10]。不同致病基因突变可导致不同WS亚型。研究表明约有15%~20%的WSII患者中检测出MITF基因突变[11]。截止到2018年10月,人类孟德尔遗传在线网的数据显示已经发现57种MITF基因杂合突变(https://www.omim.org/),大多数为错义突变和点突变,其次是移码突变、剪切位点突变和小的缺失突变。这些突变主要分布在MITF基因第6~9号外显子附近,其中点突变主要分布在7、8号外显子,对应于MITF蛋白的bHLH-Zip结构域。Tas-sabehji在1994年通过对一个WS2大家系的研究发现MITF突变,并第一次克隆出了人类MITF基因[12]。MITF是一种含有螺旋-环-螺旋碱性亮氨酸拉链(bHLH-Zip)结构的转录因子,MITF通过bHLH-Zip结构域与其他蛋白相互作用来协调靶基因的表达[13]。MITF可以促进神经嵴的黑色素细胞的分化,因此MITF突变能在许多物种中导致皮肤着色异常[14]。在内耳中,来自神经嵴的黑色素细胞存在于前庭和耳蜗。虽然他们在前庭的作用还未知,但是在耳蜗中,他们发挥着重要的作用。MITF基因突变导致耳蜗血管纹黑色素细胞的缺失,导致耳蜗内淋巴液钾离子浓度降低,内淋巴液钾离子浓度的降低会影响毛细胞对声音的反应,最终产生耳聋[15]。
该Waardenburg综合征家系中的三位WSII型患者,先证者通过高通量测序发现MITF(c.397G>T)突变,后续经一代测序验证,其母亲、外公同样存在MITF(c.397G>T)突变。先证者、其母亲、外公均有MITF基因第4个外显子c.397G>T杂合突变,编码区第397号核苷酸由鸟嘌呤变异为胸腺嘧啶,该突变极可能是终止大突变,在HGMD专业版数据库中未见报道。报告中的MITF基因转录本NM_000248,Coding Excon(即外显子长度,只包含外显子,不包含UTR和内含子)长度1269bp,共编码422个氨基酸,先证者氨基酸变化p.E133X,由原来的编码422个氨基酸,该突变极可能是终止大突变,最终只能编码132个氨基酸,导致290个氨基酸无法编码表达,极大的影响了蛋白的表达。该家系中先证者外公将致病基因传给了先症者母亲,其母亲又将致病基因遗传给了先证者。先证者母亲、外公均、先证者均为MITF基因杂合突变,并出现WSII症状,符合常染色体显性遗传的特点,有50%的风险将该致病基因遗传给后代,再生育耳聋患者的风险极高,建议行产前诊断。先证者左耳进行了人工耳蜗植入术,康复训练3月左右唤其姓名能做出反应,目前已经将近2年的言语康复训练,能与老师或其密切接触者进行一些简单的互动,可以说出2-3字简单词汇。
本研究通过NGS对WSII家系进行基因检测,发现MITF(c.397G>T)突变,该突变极可能是终止大突变,在HGMD专业版数据库中未见报道,为国际首次报道,丰富了MITF致病突变谱系。
猜你喜欢
中华实用诊断与治疗杂志(2022年1期)2022-08-31
中华实用诊断与治疗杂志(2022年1期)2022-08-31
临床输血与检验(2022年3期)2022-06-22
国际检验医学杂志(2022年2期)2022-02-11
中医眼耳鼻喉杂志(2021年1期)2021-07-22
中医眼耳鼻喉杂志(2021年2期)2021-07-21
中国药学药品知识仓库(2021年18期)2021-02-28
诊断学(理论与实践)(2020年1期)2020-04-28
水产科学(2020年2期)2020-03-20
森林工程(2018年1期)2018-05-14
