
2013—2017 年我国小反刍兽疫病毒H 基因分子演化特征
2019-09-09 13:10刘春菊盛琰翔王清华迟田英王英丽马洪超王志亮包静月
中国动物检疫 2019年9期
刘春菊,盛琰翔,王清华,迟田英,朱 琳,王英丽,马洪超,王志亮,包静月
(1. 中国动物卫生与流行病学中心,山东青岛 266032;
2. 浙江农林大学,浙江临安 311300)
小 反 刍 兽 疫(peste des petits ruminants,PPR) 是 由 小反 刍 兽 疫 病 毒( peste des petits ruminants virus,PPRV)引起的一种急性病毒性传染病,主要感染山羊、绵羊以及野生小反刍兽,以发热,眼、鼻分泌物增多以及胃炎、腹泻和肺炎为特征。该病主要流行于亚洲和非洲。2007 年我国在西藏阿里地区首次发现该病,2010 年8 月在阿里地区日土县再次发现PPR 疫情[1]。2013 年末,该病传入我国新疆地区,并迅速传播至全国22 个省份,对我国养羊业造成严重危害[2-3]。通过疫苗免疫和移动控制等措施,该病目前在我国已得到有效控制。
PPRV 属于副黏病毒科(Paramyxovirdae)麻疹病毒属(Morbolivirus);基因组为单股负链RNA,长度为15 948 nt 或者15 954 nt;基因组3'末端为前导序列(leader),5'末端为尾随序列(trailer);6 个基因排列顺序为3'-N-P-M-FH-L-5',依次编码6 个结构蛋白:核衣壳蛋白(N)、磷蛋白(P)、基质蛋白(M)、融合蛋白(F)、血凝蛋白(H)和大蛋白(L),其中P 基因还编码2 个非结构蛋白C 和V。根据F 基因或N 基因部分核苷酸序列差异,将全球PPRV 毒株分为4个基因系[4-5],其中在亚洲地区流行的毒株属于基因4 系[1]。
PPRV H 基因核苷酸长度为1 830 nt,编码609个氨基酸,编码的H 蛋白分子质量约为68.8 ku。H 蛋白和F 蛋白共同组成病毒囊膜糖蛋白。H 蛋白属于II 型膜糖蛋白,以二硫键相连的二聚体或四聚体形式存在于病毒囊膜表面。H 蛋白负责与细胞表面受体结合,然后协助F 蛋白介导病毒和宿主细胞膜的融合[6]。成熟的H 蛋白由一个短的N 端胞质侧尾巴、一段疏水的跨膜区和一个大的C 端胞外结构域组成。H 蛋白的胞外结构域构象为一个柄和一个球状头部,球状头部含有抗原表位,能够刺激机体产生中和抗体[7]。H 蛋白同时还具有神经氨酸酶活性和血凝素活性。
本研究对2013—2017 年我国PPRV 流行毒株H 基因进行序列测定和分析,探讨该病毒流行过程中的H 基因序列变异情况,掌握其分子演化特征,以期为该病控制和消灭策略的制定提供数据支持。
1 材料与方法
1.1 病毒来源
2014—2017 年 在9 个 省12 个 疫 点 采 集 的PPRV 阳性组织样品,由中国动物卫生与流行病学中心保存并提供,具体样品信息见表1。
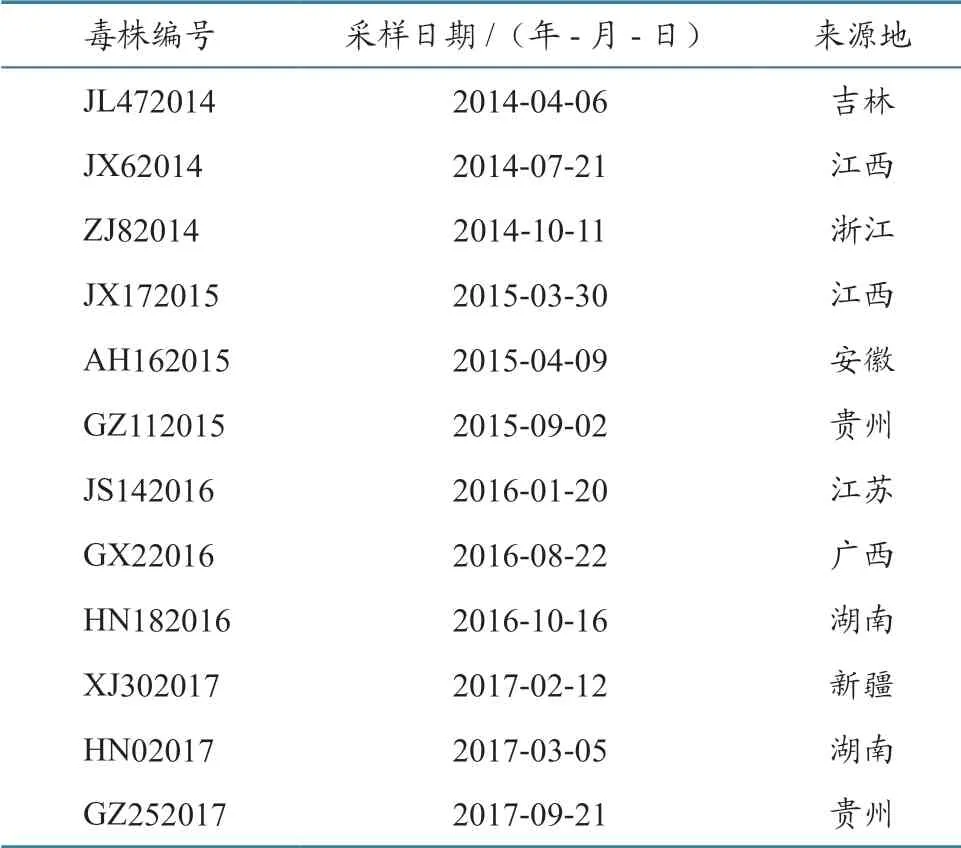
表1 2014—2017 年我国12 株PPRV 毒株信息
1.2 病毒RNA 提取
取肠系淋巴结组织100 mg,用组织匀浆仪匀浆后,根据Roche 公司生产的High Pure Viral RNA Kit 的操作说明,提取病毒RNA,-80 ℃保存。
1.3 RT-PCR
采用TaKaRa 公司生产的Prime ScriptTMOne Step RT-PCR Kit,对PPRV H 基因进行扩增:在50 μL 反应体系中加入2×One Step Buffer 25 μL,Primerscript 1 step Enzyme Mix 2 μL,上、下游引物各2 μL,病毒RNA 5 μL。RT-PCR 反应条件为:50 ℃逆转录30 min,94 ℃ 2 min,进行Taq 酶激活;40 个循环的PCR(94 ℃ 1 min,68 ℃ 1 min,72 ℃ 4 min);72 ℃ 7 min 再延伸。对PCR 产物进行切胶回收。
1.4 序列测定和进化树绘制
将纯化后的PCR 产物送青岛华大公司进行测序,获得我国12 个毒株的H 基因序列。2013—2014 年我国25 个PPRV 流行毒株的基因组序列由中国动物卫生与流行病学中心测得并已提交GenBank,毒株相关信息见表2。从GenBank 下载我国西藏及其他国家的15 个PPRV 代表毒株基因组序列,毒株相关信息见表3。从上述病毒基因组序列中截取H 基因序列。
用MEGA 4.0 软件进行序列比对,用最大似然法法构建分子进化树,bootstrap 测试1 000 次重复。用Simplot 软件进行序列同源性分析。
2 结果
2.1 H 基因核苷酸序列比较
2013—2017 年来自我国21 个省份37 个疫点的37 株PPRV 毒株H 基因核苷酸序列之间的遗传距离为0~0.007 7。其中,2013 年12 月—2014 年6 月,15 个毒株的H 基因序列完全一致。这些毒株分别来自新疆(XJ22013、XJ32013、XJ42013)、重庆(CQ2014)、黑龙江(HLJ2014)、吉 林(JL2014)、 陕 西(SaX2014)、 云 南(YN2014)、江苏(JS2014)、湖北(HB2014)、河 南(HNS2014)、 贵 州(GZ2014)、 湖 南(HN2014)、浙江(ZJ2014)、四川(SC2014)等13 个省(自治区、直辖市)。2013 年12 月—2014 年4 月,来自新疆(XJ52013)、甘肃(GS2014)、辽宁(LN2014)、安徽(AH2014)、山西(SX2014)的5 个毒株H 基因序列完全一致。毒株XJ302017 与HN02017 之间的H 基因核苷酸序列差异最大,遗传距离为0.007 7,共14 个位点存在序列差异。
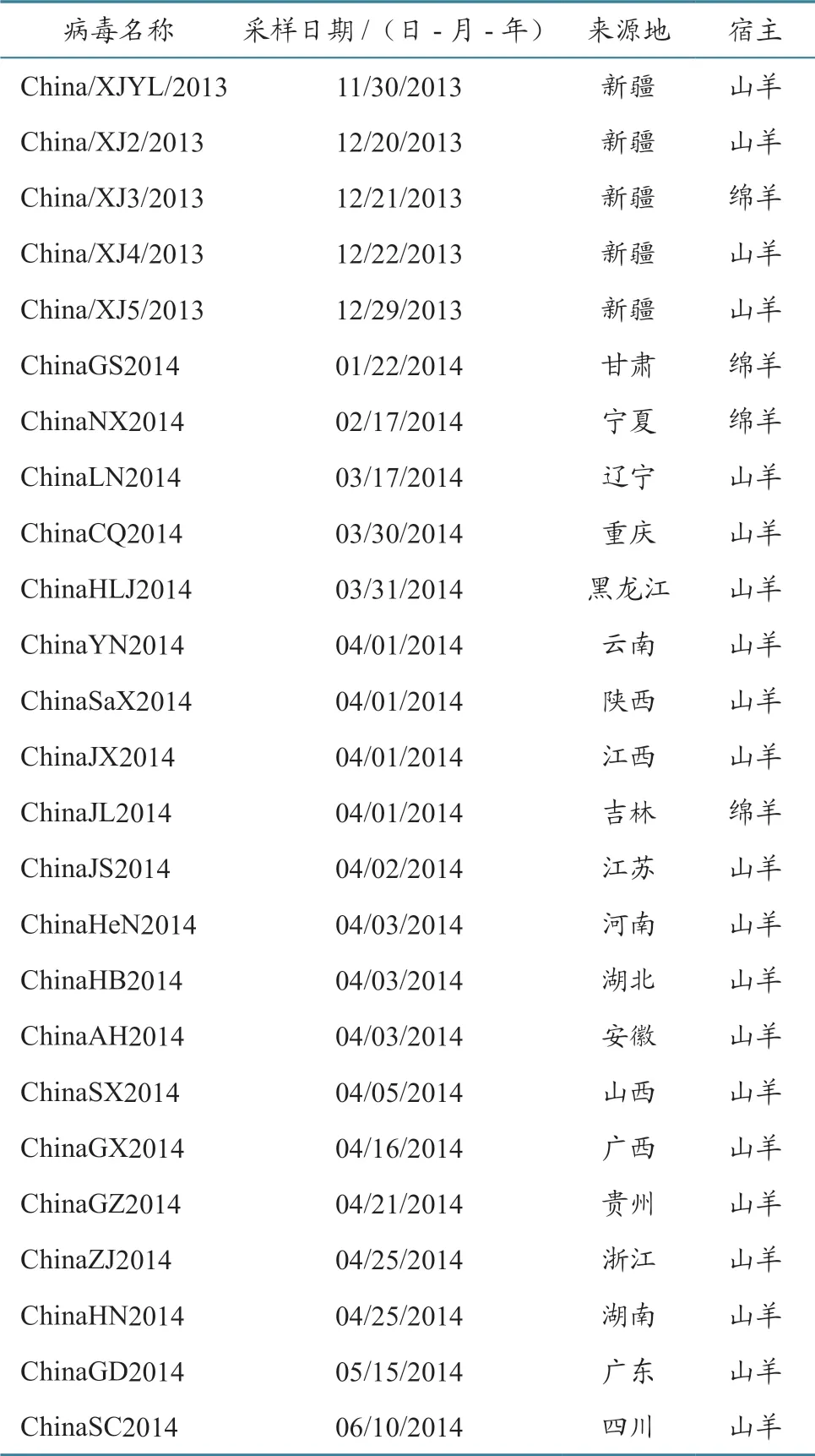
表2 2013—2014 年我国PPRV 流行毒株信息
37 个毒株之间的核苷酸变异分布于H 基因的41 个位点。Simplot 分析结果(图2)显示,H 基因序列中,120~420 bp 间的序列同源性为100%,没有突变发生。H 基因核苷酸序列在3 个区域变异较大,分别为680~820 bp、1 240~1 380 bp 和1 680~1 720 bp 区域。
H 基因41 个核苷酸变异位点中,24 个导致了氨基酸序列的改变(表4):18 个变异发生于第1位密码子,导致其中的15 个位点发生氨基酸改变;9 个变异位于第2 位密码子,导致全部氨基酸发生改变;14 个变异发生于第3 位密码子,未导致氨基酸改变。

表3 PPRV 参考毒株信息
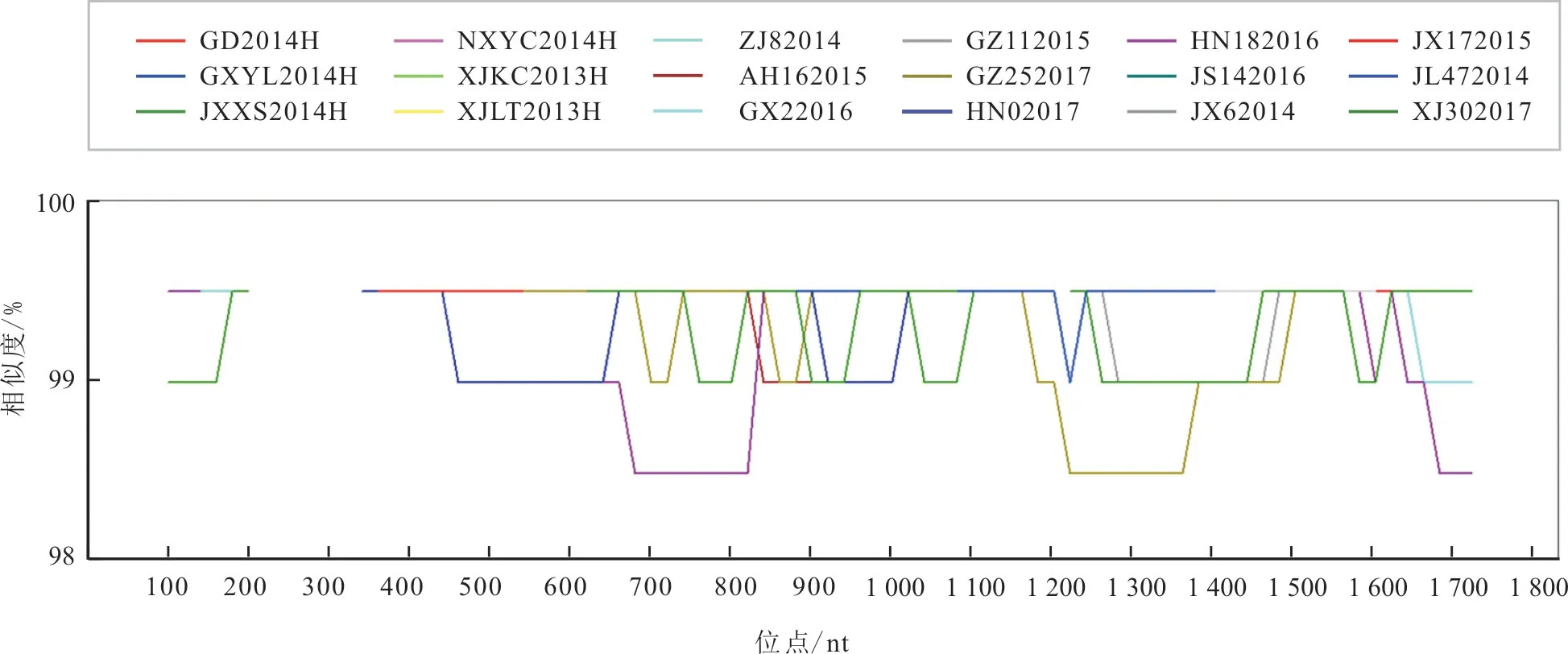
图1 2013—2017 年我国PPRV 毒株H 基因核苷酸序列同源性Simplot 分析结果
将XJYL2013 株与其余36 个毒株进行H 基因核苷酸序列比对,发现36 个毒株分别在1~9 个位点发生了突变。H 基因核苷酸序列突变位点数与毒株流行时间呈正相关,2016 年10 月以前的32 个毒株,突变位点为1~6 个,2016 年10 月以后的4个毒株,突变位点为8~9 个。
2.2 H 基因编码氨基酸序列比较
2013—2017 年我国37 株PPRV 毒株H 基因编码氨基酸序列之间的遗传距离为0~0.013 2。H基因编码的609 个氨基酸位点中,24 个位点发生了突变:胞内区(1~34 位aa)有3 个位点发生了突变,跨膜区(35~58 位aa)有1 个位点发生突变,胞外长柄区(59~181 位aa)有3 个位点发生突变,胞外球状头部区(182~609 位aa)有17 个位点发生突变。
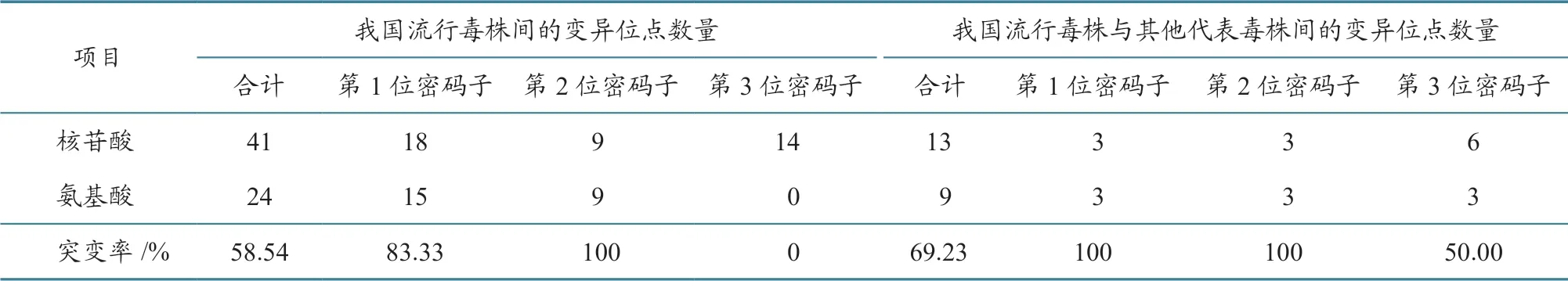
表4 2013—2017 年我国PPRV 毒株H 基因核苷酸和编码氨基酸序列变异情况
2.3 H 基因序列差异分析
2013—2017 年我国37 株PPRV 流行毒株与15 株PPRV 代表毒株的H 基因核苷酸序列遗传距离为0.019 5~0.146 9,其中与2007—2008 年我国西藏地区流行毒株H 基因核苷酸序列遗传距离为0.019 5~0.025 7,而西藏地区流行毒株之间的H基因核苷酸序列遗传距离为0.000 5~0.002 7。与15 株PPRV 代表毒株进行序列比对,发现2013—2017 年我国37 株PPRV 流行毒株H 基因的13 个位点发生了核苷酸序列突变,其中9 个导致了氨基酸序列的改变(表4):3 个变异位于第1 位密码子,3 个变异位于第2 位密码子,全部导致氨基酸改变;6 个变异位于第3 位密码子,3 个导致氨基酸改变。
2013—2017 年我国37 株PPRV 流行毒株与15 株代表毒株H 基因编码氨基酸序列的9 个变异位点位于179~591 aa 之间,全部位于H 蛋白的胞外区,其中8 个变异位点位于胞外球状头部区。
2.4 系统发育树构建
基于H 基因核苷酸序列构建系统发育树,发现2013—2017 年我国37 株PPRV 流行毒株构成基因4 系中一个独立的进化小分支,而我国西藏地区流行毒株与印度2014 年毒株形成一个小分支(图2)。
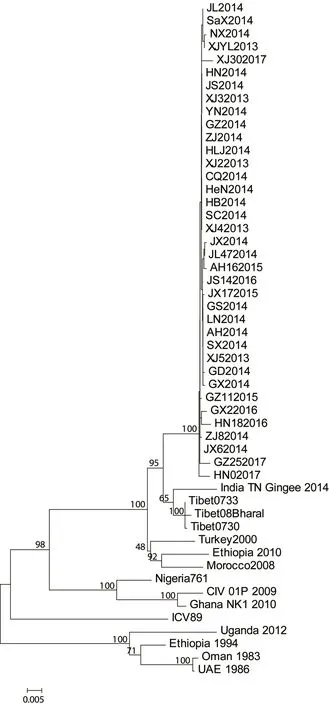
图2 PPRV 毒株H 基因分子进化树
3 讨论
本研究对2013—2017 年我国21 个省份37 个疫点的PPRV 流行毒株H 基因进行序列分析,探明了PPRV 流行过程中H 基因序列变异情况,为PPR 控制和消灭策略的制定提供了数据支持。
2013—2017 年我国37 个PPRV 流行毒株的H 基因变异较大。PPRV 在我国流行的4 年间,H基因的41 个核苷酸位点发生了突变,其中24 个位点导致了氨基酸序列的改变,非同义突变率为58.5%。之前的研究[8]发现,2013 年11 月—2014年6 月,25 个毒株H 基因的7 个位点发生突变,其中4 个位点导致氨基酸序列的改变,非同义突变率为57.1%。本研究和之前的研究都发现,H 基因的非同义突变主要由第1 位和第2 位密码子的突变引起。PPRV 在我国流行过程中,其突变在H 基因中随机分布,突变位点数量与流行时间呈正相关。因此,持续实时监测PPRV H 基因分子变异,对于该病的防控具有重要意义。
2013—2017 年我国流行的PPRV 毒株在分子进化树中聚类为一个独立小分支,相比于其他PPRV 代表毒株,这些毒株的H 基因在13 个位点发生了特征性的核苷酸突变,其中9 个位点导致了氨基酸序列的改变,而且这些突变的氨基酸位点主要位于胞外区。作为重要的抗原蛋白,H 蛋白的胞外球状头部含有抗原表位,能够刺激机体产生中和抗体。对于我国PPRV 流行毒株H 蛋白氨基酸改变能否导致其抗原性改变,还需进一步研究。
猜你喜欢
动物医学进展(2022年9期)2022-11-26
世界科学技术-中医药现代化(2022年3期)2022-08-22
肝博士(2022年3期)2022-06-30
科学大观园(2022年2期)2022-01-23
文萃报·周二版(2021年47期)2021-12-14
福建农业学报(2021年6期)2021-08-18
教学考试(高考生物)(2020年4期)2020-11-18
发明与创新·中学生(2019年6期)2019-06-26
安徽农业科学(2018年1期)2018-05-14
