
高温下季戊四醇结构和导热率的分子动力学研究
2019-09-10 07:22宫薛菲杨启容姚尔人李灿
青岛大学学报(工程技术版) 2019年2期
宫薛菲 杨启容 姚尔人 李灿



摘要: 针对季戊四醇材料在储存和使用方面存在的优势,本文以季戊四醇为研究对象,采用Materials Studio软件,通过分子动力学模拟手段,对季戊四醇进行建模和结构优化,研究了不同温度对季戊四醇分子结构和导热率的影响,分析了分子键长键角随温度变化的趋势。研究结果表明,温度对C—C键长影响最大,对C—O—H键角影响最大,固固相变发生时,C—O键长和O—C—H键角发生急剧变化,说明温度对季戊四醇分子部分键长键角的变化具有一定影响,特别是固固相变温度附近变化幅度更加明显;当温度在430~480 K时,季戊四醇导热率在0.67 W/(m·K)左右,说明导热率在模拟温度范围内,随温度升高而增大,固固相变对导热率的变化影响不大。该研究对季戊四醇作为储能材料在实际应用中具有参考意义。
关键词: 季戊四醇; 分子动力学模拟; 键长; 键角; 导热率
中图分类号: TK124; O357.5+3 文献标识码: A
季戊四醇是一种典型的新戊基多元醇类,其化学名称是2,2二羟甲基1,3丙二醇[1],主要用来生产润滑剂、醇酸树脂、聚氨酯、表面活性剂、乳化剂及炸药等[2],在树脂、涂料、化工、医药及国防等领域广泛应用[3]。季戊四醇的碳链较长,分子量较高,醚键稳定[4],因其高度对称性的分子结构(体心立方结构,分子中含有4个等同的羟甲基)所形成特有的性质[5],并且季戊四醇材料在晶型转变时能可逆地吸收和释放大量的相变潜热[6],因此许多学者对其作为固固相变的一种储能材料进行不断深入的研究[7]。相变储能材料是利用分子自身形态或分子排序的改变,使大量热量在相变过程中被吸收或释放,从而实现控制温度和储存能量的作用[8]。相变储能材料的储能密度高、充放热过程中温度变化较小,因此受到国内外学者的广泛关注[9]。固固相变储能材料在相变过程中没有液相的产生[10],体积变化较小,无毒,无腐蚀,对容器的材料和制造技术要求不高,使用寿命长[11],具有较好应用前景[12]。逯来玉等人[13]对不同温度下季戊四醇的结构和振动性质进行了分子动力学模拟,指出了键长键角随温度的变化趋势,说明了温度对分子结构的影响;陈占秀[14]对丙三醇/1.6己二醇混合体系进行相变模拟,指出了自扩散系数和比体积可作为固液相变的判断依据;闫全英等人[15]对季戊四醇的挥发性进行研究,说明在固固相变材料使用过程中需要考虑封装问题;Y.Takahashi等人[16]通过实验测定季戊四醇浆液的比热容和导热率等热物性参数,给出较为准确的实际数据;陈爱英等人[17]认为季戊四醇的固固相变温度为460 K左右;叶振强等人[18]对热导率的分子动力学模拟方法进行了对比研究。但对季戊四醇固态导热率的研究还相对较少。因此,本文以Materials Studio软件为基础,建立了季戊四醇体系模型,在不同温度下对其进行分子动力学模拟[19],研究温度对分子结构的影响,计算温度在460 K左右季戊四醇的导热率。该研究具有广泛的应用前景。
1 模拟与计算
1.1 模型建立
季戊四醇常温下为白色粉末状晶体,其结构式为C(CH2OH)4,是典型的星型结构,季戊四醇分子结构如图1所示。Materials Studio是一款为材料学研发的PC软件,既可以使用分子库提供的分子模型,也可以自定义想要的分子模型,为分子动力学模拟提供了很好的模型基础。
首先在Materials Studio软件中新建一个project目录,然后新建3D Atomistic Document文件,运用绘制工具栏绘制季戊四醇单体,调整氢键位置,通过Clean功能项优化,建立体心立方结构的季戊四醇單体。季戊四醇单体分子如图2所示。运用Amorphous Cell中的Construction功能项,在298 K常温下,以96个单体(2 016个原子)填充晶胞,查阅数据库得知,季戊四醇密度为1.399 g/cm3,构建季戊四醇晶胞模型,晶胞尺寸由密度决定,该研究所做的诸多模拟,均以此模型为模拟对象。季戊四醇晶胞构型如图3所示。
1.2 结构优化
如果初始构型离平衡态非常远,体系受力可能过大,导致MD模拟失败。针对于此,通过Discover模块中的Minimizer功能项进行能量最小化处理,但体系仍可能存在势能面上局部极小值之间的能垒,所以进一步通过Forcite模块中的“退火处理”对体系进行结构优化。设定模拟的起始温度为300 K,然后每30 K升温1次,升温后平衡计算的时间设定为20 ps,直至升到终止温度420 K,再进行降温处理,每30 K降温1次,直到降至起始温度,结构优化的整个过程在恒温恒压(normal pressure and temperature,NPT)系统下进行,通过升温再降温这一“退火处理”过程,可以尽量消除模型中的局部不合理结构。
2 结果与分析
2.1 键长
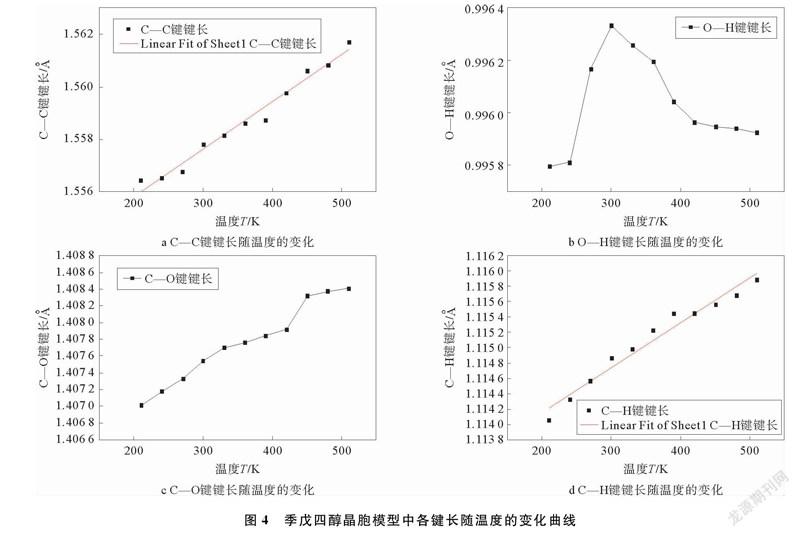
季戊四醇晶体模型中存在384个C—C键,384个O—H键,384个C—O键和768个C—H键,取多次模拟后数值的平均值做出各键长随温度变化的曲线。季戊四醇晶胞模型中各键长随温度变化曲线如图4所示。
由图4可以看出,C—C键、C—O键、C—H键的键长都随温度的升高而逐渐增加,键变得越脆弱,越容易断裂,这与常识相吻合,而O—H键键长随温度的变化曲线是一个例外,其变化趋势以300 K为界,当温度小于300 K时,随着温度升高,键长增加,当温度大于300 K时,随着温度的升高,键长减小。由计算结果可以看出,C—C键的键长增加幅度为0.34%,O—H键的键长变化幅度为0.05%,C—O键的键长增加幅度为0.1%,C—H键的键长增加幅度为0.16%,所以在210~510 K温度范围内,C—C键的变化幅度最大,尤其是C—O键的键长在420~450 K区间内发生急剧变化,说明固固相变时,部分键长发生大幅度变化,这与能量的吸收与释放有关,对分子结构有一定影响。
2.2 键角
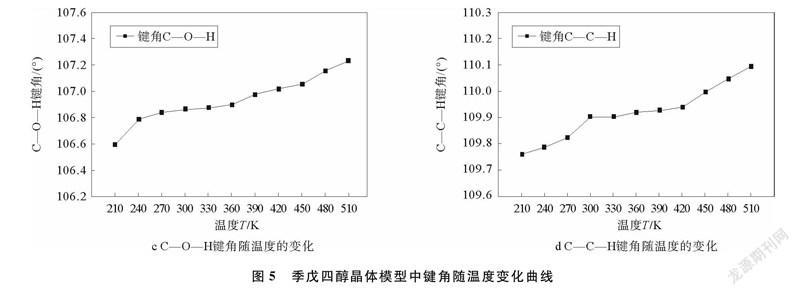
在季戊四醇晶体模型中,存在576个C—C—C键角,768个O—C—H键角,384个C—O—H键角和768个C—C—H键角,取其多次模拟后结果的平均值,做出各键角随温度变化曲线,季戊四醇晶体模型中键角随温度变化曲线如图5所示。
由图5可以看出,键角C—C—C和键角O—C—H随着温度的升高而减小,而键角C—O—H和键角C—C—H则随着温度的升高而增大。由计算结果可以看出,键角C—C—C和键角O—C—H分别减少了0.17%和0.09%,而键角C—O—H和键角C—C—H则分别增加了0.6%和0.3%,所以温度对键角C—O—H和键角C—C—H的影响比较大,特别是键角O—C—H的大小在420~450 K区间内发生急剧变化,同样反映了固固相变对部分键角的变化产生较大影响。
2.3 导热率
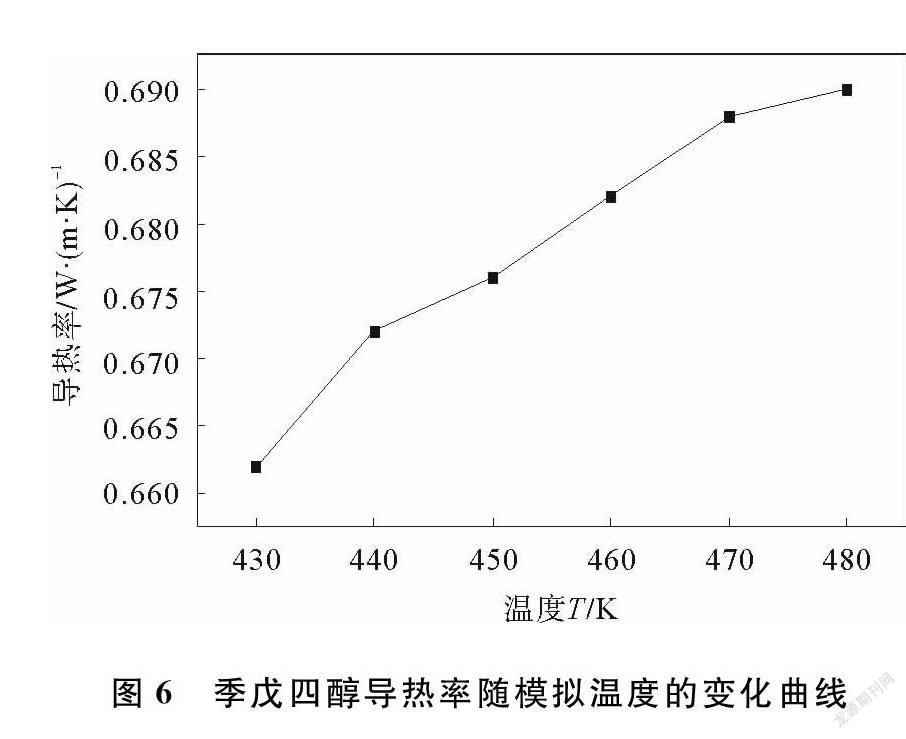
理论上,模拟时间越长,导热率越稳定,计算结果越精确,通过数据整理,可得季戊四醇导热率随模拟温度的变化曲线如图6所示。由图6可以看出,随着温度的升高,季戊四醇的导热率逐渐增大,但是在其固固相变温度左右,曲线斜率并没有发生大幅度变化,即导热率没有发生较大变化,这是由于固固相变发生时,没有产生固液相态的变化,对季戊四醇的导热率没有产生较大影响。
3 结束语
本文主要研究了温度对季戊四醇结构的影响,通过季戊四醇分子键长和键角的变化,反映了温度的影响,计算季戊四醇在固固相变温度附近的导热率。通过对模拟数据的分析可知,温度对C—C键键长影响最大,对C—O—H键角影响最大,固固相变发生时,C—O键键长和O—C—H键角发生急剧变化,说明固固相变的发生对分子结构造成比较明显的影响。通过模拟计算,可得,430~480 K的季戊四醇导热率在0.67 W/(m·K)左右,导热率随温度的增加而增大,说明导热率受固固相变的影响不大,数值上没有发生剧变。本研究是建立在分子动力学模拟的基础之上,并没有辅以实验作对比,但对实验研究具有一定的参考意义,今后會在模拟的基础上,通过实验数据加以对比,探究季戊四醇及其混合物的热力学性质,在相变储能材料领域具有更实际的应用价值。
参考文献:
[1] 扈士海. 季戊四醇的生产应用及市场分析[J]. 河北工业科技, 2009, 26(1): 5860.
[2] Hesse A, Biyikal M, Rurack K, et al. Development of highly sensitive and selective antibodies for the detection of the explosive pentaerythritol tetranitrate (PETN) by bioisosteric replacement[J]. Journal of Molecular Recognition Jmr, 2016, 29(2): 8894.
[3] 肖铭. 我国季戊四醇生产技术进展及市场分析[J]. 精细与专用化学品, 2017, 25(11): 2025.
[4] Shiono R, Cruickshank D W J, Cox E G. A refinement of the crystal structure of pentaerythritol[J]. Acta Crystallographica, 2010, 11(6): 389391.
[5] 陈春霞. 季戊四醇生产技术进展及市场分析[J]. 精细与专用化学品, 2010, 18(1): 813.
[6] 赵建国. 季戊四醇的研制与应用[J]. 辽宁化工, 2011, 40(1): 8082.
[7] Wang X P, Sun Y J, Sun K K. Experimental investigation for the solubility of R1234ze(E) in pentaerythritol tetrahexanoate and pentaerythritol tetraoctanoate[J]. Fluid Phase Equilibria, 2015, 400(25): 3842.
[8] Pielichowska K, Pielichowski K. Phase change materials for thermal energy storage[J]. Progress in Materials Science, 2014, 65(10): 67123.
[9] 冷光辉, 曹惠, 彭浩, 等. 储热材料研究现状及发展趋势[J]. 储能科学与技术, 2017, 6(5): 10581075.
[10] Yuan K J, Zhang Z G, Fang X M, et a1.Research progress of inorganic hydrated salts and their phase change heat storage composites[J]. Chemical Industry and Engineering Progress, 2016, 35(6): 18201826.
[11] Ge Z W, Ye F, Lasfargues M, et a1. Recent progress and prospective of medium and high temperatures thermal energy storage materials[J]. Energy Storage Science and Technology, 2012, 1(2): 89102.
[12] Zhang H L, Fang X, Zhao Y J. Progress in phase change materials and technologies[J]. Materials Review, 2014, 28(7): 2632.
[13] 逯来玉, 周晓林, 姬广富, 等. 高温下季戊四醇晶体的结构和振动性质的分子动力学模拟[J]. 原子与分子物理学报, 2012, 29(5): 908912.
[14] 陈占秀. 相变储能材料相变过程的分子动力学研究[D]. 天津: 天津大学, 2012.
[15] 闫全英, 王威. 在储能过程中多元醇类相变材料挥发性的实验研究[J]. 材料导报, 2005, 19(s2): 269270.
[16] Takahashi Y, Kamimoto M, Abe Y, et al. Heat capacity, heat of transition, and thermal conductivity of pentaerythritol and its slurry[J]. Jpn. J. Thermophys. Prop, 1988, 2(1): 5358.
[17] 陈爱英, 汪学英. 相变储能材料及其应用[J]. 洛阳理工学院学报: 自然科学版, 2002, 12(4): 79.
[18] 叶振强, 曹炳阳, 李元伟. 热导率的平衡分子动力学模拟中的热流计算[J]. 计算物理, 2015, 32(2): 186194.
[19] Bordat P, Marbeuf A, Brown R. Molecular simulation of solidsolid phase transitions[J]. Molecular Simulation, 2006, 32(12): 971984.
