
线粒体自噬的调控机制及其在相关疾病中的作用
2019-10-14 02:44林晶晶杨宇丰
生物技术进展 2019年5期
林晶晶, 杨宇丰
福州大学生物科学与工程学院, 福州 350108
自噬(autophagy)是指通过不同途径向溶酶体传递并降解细胞成分(如细胞质、细胞器和蛋白质聚集物等),这些细胞组分被隔离并包裹至一个双层膜结构,这种结构被称为自噬体(autophagosome)[1]。在酿酒酵母的遗传学筛选中已发现了30多种自噬相关基因(autophagy-related gene,Atg),负责调控整个自噬过程和参与自噬发生的不同阶段,这些基因在酵母和哺乳动物中高度保守。自噬缺陷会导致细胞损伤的积累,与肝病、神经退行性病变、衰老、癌症和代谢综合征等密切相关[2]。
根据机制和作用的不同,可将自噬分为3种类型:巨自噬、微自噬和分子伴侣介导的自噬,其中巨自噬是最主要的自噬形式。巨自噬可进一步分为选择性巨自噬和非选择性巨自噬,而线粒体自噬就是选择性巨自噬的一种,是指通过特异性降解细胞内受损的或者多余的线粒体并循环利用其组成元素的过程。线粒体自噬最初是由Kim等[3]通过早期的电镜研究在哺乳动物细胞中发现的,线粒体被包裹到带自噬标记物MAP1轻链3(microtubule-associated protein 1 light chain 3,LC3)的囊泡中。LC3是酵母中Atg8的同源物,是一种类泛素蛋白,在自噬发生过程中与磷脂酰乙醇胺共价连接参与形成隔离膜[4]。线粒体自噬是一个缜密有序的巨自噬过程,利用了巨自噬的相关元件,因此,线粒体自噬从酵母到哺乳动物也都高度保守。线粒体自噬分为5个阶段:隔离膜(isolation membrane)的形成、隔离膜延伸成为自噬前体(phagophore)并靶向到线粒体上、边缘封闭隔离线粒体形成自噬体、自噬体通过细胞骨架系统与溶酶体(lysosome)融合形成自噬溶酶体(autolysosome)、受损或多余的线粒体随后降解。线粒体作为细胞的能量工厂,除了生产ATP,还在细胞凋亡、血红素和类固醇合成、Ca2+调节等过程中发挥关键作用,其功能正常与否对细胞本身至关重要。线粒体也是一个动态的网络状细胞器,通过不断地改变其形态和功能以适应细胞的需要。同样,受损或多余线粒体的有效清除也是维持细胞内环境稳定的关键。异常的线粒体自噬与神经退行性疾病、癌症、肌肉性疾病、糖尿病、肥胖和心脏病等都有关[5]。
自线粒体自噬概念被提出以来,有关线粒体自噬途径的研究得到了广泛的关注,其中,PINK1/Parkin途径是目前研究较多的参与哺乳动物线粒体自噬的经典通路。帕金森症(Parkinson’s disease,PD)是第二大常见的神经退行性疾病,发病率仅次于阿尔海默兹症(Alzheimer’s disease,AD),多种PD相关基因被发现突变能导致PD的发生,PINK1和Parkin是其中较早被发现的与常染色体隐性遗传早发性PD相关的致病基因。线粒体稳态的维持对于机体健康至关重要,线粒体自噬功能障碍与PD等疾病的发生密切相关,其功能异常增高也同样会导致疾病。本文主要讨论了酵母和哺乳动物内正向调控线粒体自噬的途径,包括经典通路PINK1/Parkin途径和其他受体介导的线粒体自噬途径,并对目前研究较少的线粒体自噬的负调控机制进行了综述,最后探讨了线粒体自噬功能异常与人类疾病(如帕金森症)的关联。目前,线粒体自噬的分子调控机制是线粒体自噬和细胞自噬研究领域广泛关注的焦点,这方面的研究对于揭示线粒体自噬相关疾病的发病机理及防治机制具有重要意义,同时也有助于寻找新的靶点,推动新药研发与临床治疗。
1 酵母中的线粒体自噬
线粒体自噬现象首先是在酵母中发现的[6],并利用酵母系统自身的优势对其分子机制进行了充分的研究。大多数Atg对于巨自噬都是必需的,也有些Atg(如Atg11)只对特定类型的选择性巨自噬是必需的[7]。线粒体外膜蛋白Atg32是酵母中线粒体自噬的特异受体,其对非选择性巨自噬、胞质至液泡途径(the cytoplasm to vacuole targeting pathway,Cvt pathway)、过氧化酶体自噬的发生是非必需的。Atg32由529个氨基酸组成,具有单一的跨膜结构——跨线粒体外膜,其N端和C端分别暴露在胞质和线粒体间质中,且含WXXI结构域[8]。
在酵母中,线粒体受损后诱导线粒体自噬,Atg32上的Ser114和Ser119位点(特别是Ser114)被磷酸化,随后被磷酸化的Atg32与Atg11互作形成复合物[9],Atg11通过与Atg8相互作用使得线粒体被自噬元件识别,随后招募到自噬前体中;Atg32还可以通过WXXI胞质结构域直接与Atg8结合,同样招募线粒体进入自噬体(图1)[10]。近期研究发现,酵母中的线粒体自噬还与磷脂生物合成途径有关,磷脂生物合成途径是磷脂酰乙醇胺通过2个甲基转移酶Cho2和Opi3转化为磷脂酰胆碱,磷脂甲基化的调控对于Atg32介导的线粒体自噬至关重要[11]。此外,在氧化条件下Atg32缺失的酵母中没有发现线粒体功能缺陷[8,12],这表明酵母中存在另一条不依赖于Atg32的线粒体自噬通路,不同的通路可能介导不同目的的线粒体自噬。
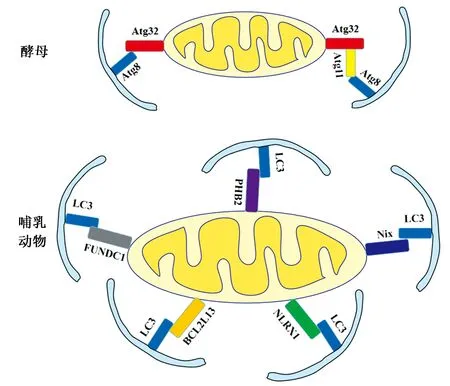
图1 酵母和哺乳动物中线粒体自噬的特异性受体Fig.1 The specific receptors of mitophagy in yeast and mammals.
2 哺乳动物中的线粒体自噬
2.1 PINK1/Parkin介导的线粒体自噬
PINK1是由PARK6基因编码的高度保守的线粒体蛋白,包含N端线粒体定位信号序列(mitochondrial targeting signal,MTS)、α螺旋跨膜结构域(transmembrane,TM)、丝氨酸/苏氨酸激酶结构域以及C端线粒体外膜滞留信号肽序列。Parkin是由PARK2基因编码的蛋白质,含有465个氨基酸。Parkin是泛素-蛋白酶体系统(ubiquitin-proteasome system,UPS)中选择性降解机制的关键,负责将泛素(ubiquitin,Ub)分子连接到底物蛋白,加上Ub标签的底物蛋白被蛋白酶体识别后降解。遗传学上的相互作用表明,PINK1与Parkin位于同一条通路上发挥保护线粒体的功能,且Parkin位于PINK1下游[13]。
在线粒体膜电势正常的情况下,PINK1可结合外膜上的TOM复合物由细胞质转移到线粒体内膜上,随后被多种线粒体蛋白酶裂解[14]。经典的PINK1/Parkin模型是线粒体受损后膜电势降低,PINK1不能转运至内膜而是聚集在去极化的线粒体外膜上,接着PINK1磷酸化Ub的Ser65,磷酸化的泛素(pSer65-Ub)募集泛素连接酶Parkin至线粒体上。这种直接或间接的募集方式是通过pSer65-Ub与细胞溶质分子的交流,向细胞质传递线粒体损伤的信息,将Parkin从胞质募集至受损线粒体外膜上[15],在这一过程中,PINK1在Ser228和Ser402位点的自身磷酸化是募集Parkin所必需的,且PINK1激酶活力与线粒体定位信号也是不可缺少的[16]。随后,PINK1通过磷酸化Parkin的Ser65激活其泛素连接酶活性,将Lys63和Lys48连接的多泛素链连接到线粒体外膜蛋白(如水通道蛋白VDAC1等)。胞质中的自噬系统接头蛋白p62和Beclin-1能结合这种泛素化蛋白,p62可以与LC3直接相互作用,从而锚定到自噬体的隔离膜上;蛋白Beclin-1可以招募Vps34-15-AMBRA复合物,与自噬体延伸元件Atg5-12/Atg16结合导致自噬体的形成。最终,线粒体自噬是通过线粒体自噬体与溶酶体融合随后降解完成的[17~20]。
线粒体融合分裂机制与线粒体自噬密切相关,膜电势正常的线粒体才能进入正常的融合分裂周期,而去极化的线粒体则走向自噬的命运[21]。PINK1和Parkin在可调控线粒体融合与分裂中也扮演着重要角色,研究表明PINK1和Parkin具有促进线粒体分裂的功能[22]。当线粒体去极化后,PINK1招募Parkin到线粒体上,Parkin泛素化线粒体融合蛋白,随后线粒体融合蛋白被蛋白酶体降解,这使得受损的线粒体无法与线粒体网络融合而是通过自噬被清除[23]。此外,PINK1和Parkin还可通过调控线粒体运动来隔离受损的线粒体。Miro是将肌动蛋白固定在线粒体表面的初级马达/适配器蛋白复合物的组成部分,线粒体去极化后,PINK1磷酸化Miro,随后被Parkin依赖的泛素蛋白酶体途径降解。Miro的降解阻滞了线粒体的运动,这有助于在去极化线粒体被自噬体吞噬之前将其隔离。通过隔离不健康的线粒体,可以将活性氧自由基(reactive oxygen species,ROS)对线粒体的损伤局限在特定的区域内,随后通过线粒体自噬清除受损的线粒体[24]。总之,哺乳动物细胞可以通过调节线粒体的清除、分布和运动来维持能量平衡和避免氧化应激,而PINK1/Parkin通路可以通过线粒体融合蛋白等相关蛋白调控线粒体的融合与分裂,也可以借助Miro调节线粒体运动来介导线粒体自噬。
2.2 Nix介导发育过程中的线粒体自噬
程序性线粒体自噬即存在于发育过程中的线粒体自噬发生在红细胞成熟过程中,目的是清除成熟红细胞中的线粒体。研究表明,位于线粒体外膜的Bcl-2家族蛋白Nix参与了这一过程,在Nix缺失的小鼠成熟红细胞中有线粒体的存在,而Nix缺陷会抑制红细胞成熟,导致贫血[25]。Nix定位于线粒体外膜,胞质部分含有WXXL结构域,可与LC3及其同源蛋白GABARAP(GABA受体相关蛋白)相结合,将线粒体锚定到自噬体的隔离膜上,且Nix依赖的膜电势对线粒体靶向到自噬体十分重要。此外,上调Nix蛋白可促进线粒体自噬抑制蛋白Bcl-2和Bcl-XL与Beclin-1的解离,从而影响自噬体的形成[26]。
Nix不仅在成熟红细胞的线粒体清除过程中起作用,还参与介导干细胞分化过程中的线粒体自噬。诱导性多能干细胞(induced pluripotent stem cells,iPSCs)主要依赖糖酵解作为能量来源,干细胞比体细胞有更少的未成熟线粒体,因此在体细胞重编程过程中,体细胞线粒体需要被清除。在体细胞的分化过程中线粒体质量先增加,随后通过线粒体自噬减少。有研究发现,Nix对转录因子Sox2、Klf4、Pou5f1/Oct4(SKP/SKO)细胞重编程中的线粒体消除至关重要,而这一过程不依赖线粒体膜电势的降低[27]。上述结果表明Nix还可能在其他发育过程中起重要作用。
2.3 FUNDC1介导缺氧诱导的线粒体自噬
FUNDC1是介导线粒体自噬的线粒体外膜蛋白,其包含3个跨膜域,以及位于N端保守的LC3结合域(LC3-interaction region,LIR)——Y(18)xxL(21)序列,此序列可在哺乳动物中与LC3互作介导缺氧引起的线粒体自噬。缺氧可导致FUNDC1去磷酸化,并增强其与LC3的相互作用,促进自噬体的形成[28]。LC3B可以识别FUNDC1位于LIR结构域的Ser17位点的磷酸化状态,表明FUNDC1磷酸化或去磷酸化是其作为线粒体自噬受体被特异识别的关键[29]。FUNDC1介导线粒体自噬不依赖Parkin,而另一线粒体E3泛素连接酶MARCH5却能通过泛素降解FUNDC1来调控缺氧状态下的线粒体自噬[30]。此外,FUNDC1可以与线粒体分裂蛋白Drp1和线粒体融合蛋白Opa1相互作用,调控线粒体分裂或融合和线粒体自噬。Opa1通过Lys70(K70)残基与FUNDC1相互作用,K70突变使得FUNDC1-Opa1复合物解体从而促进线粒体分裂和线粒体自噬;FUNDC1的去磷酸化促进了FUNDC1与Opa1解离并与Drp1的结合,同样促进线粒体自噬。总之,FUNDC1既调节线粒体分裂或融合,又调节线粒体自噬[31]。
2.4 新型线粒体自噬受体
除了上述3种调控线粒体自噬的途径外,还有几种新发现的线粒体自噬受体能够介导线粒体的特异清除。Bcl-2类蛋白13(BCL2-like 13,BCL2L13)是新发现的Atg32功能性同源蛋白,参与介导线粒体自噬和线粒体分裂。BCL2L13表达并定位于线粒体外膜上,包含1个C端单跨膜结构域、4个保守的BCL2同源结构域(BCL2 homology domains,BH1-4)以及位于147~150和273~276位点的2个WXXL/I基序。其中,BH1-4结构域与BCL2L13诱导的线粒体片段化有关。位于273~276位点上的第2个WXXL/I基序是一个可以与LC3结合的LIR,因此,BCL2L13可将LC3招募到线粒体表面,导致线粒体自噬小体的形成从而介导线粒体自噬。BCL2L13对于线粒体损伤引起的碎片化和线粒体自噬是必不可少的,诱导和/或磷酸化BCL2L13可以调节其活性,且不依赖线粒体分裂蛋白Drp1和E3泛素连接酶Parkin。此外,BCL2L13还能诱导Atg32缺陷酵母中的线粒体自噬,表明其功能具有保守性[32]。
NLRX1也是一个新发现的线粒体自噬受体,该受体介导了李斯特菌(Listeriamonocytogenes)感染诱导的线粒体自噬。NLRX1是Nod样受体(the only Nod-like receptor,NLR)家族的成员,含有MTS和LIR结构域。L.monocytogenes可通过诱导巨噬细胞的线粒体自噬来逃避宿主细胞的杀伤,具体机制是L.monocytogenes通过产生溶血素O(listeriolysin O,LLO)诱导NLRX1的寡聚化,促进LIR结构域与LC3的结合,进而诱导线粒体自噬[33]。此外,线粒体内膜蛋白Prohibitin 2(PHB2)也是重要的线粒体自噬受体。线粒体去极化后,蛋白酶体介导的线粒体外膜破裂会暴露线粒体内膜上的PHB2,而PHB2上含有LIR结构域,与LC3相互作用后介导线粒体与自噬体的结合。PHB2是哺乳动物中Parkin介导的线粒体自噬所必需的,同时参与线虫中胚胎受精后父系线粒体的清除[34]。
综上所述,除了PINK1/Parkin介导的线粒体自噬的调控机制较为清楚,其他机制(包括Nix、FUNDC1、BCL2L13、NLRX1、PHB2)介导的调控还有待进一步研究。这些受体蛋白质都具有1个LIR结构域,能通过LIR与LC3相连,定位到自噬小体上并进行后续的延伸及融合等步骤(图1)。上述线粒体自噬通路的发现为研究哺乳动物细胞的线粒体质量控制提供了更多的保障。
3 介导负调控线粒体自噬的途径
异常或过度的线粒体自噬对机体是有害的,因此需找出一个平衡点,使胞内的线粒体自噬保持在最佳水平以适应外界压力负荷的变化。而这种平衡就需要通过多种机制调控,包括已知的PINK1、Parkin、Nix等参与的在生理水平下正调控线粒体自噬,以及研究较少的负调控机制。
3.1 Parkin/USPs介导的可逆泛素化过程负调控线粒体自噬
目前关于线粒体自噬的负调控机制的研究主要是围绕在以泛素连接酶Parkin和泛素特异蛋白酶(ubiquitin-specific protease,USPs)为中心的可逆泛素化过程。线粒体去极化诱导Ub的Ser65磷酸化后激活Parkin泛素连接酶活性,在线粒体上Lys6、Lys11和Lys63位点连接泛素链后,线粒体定位的USPs会剪切Lys6、Lys11、Lys63及Lys48(特别是Lys6和Lys11)连接的多聚体。其中,USP15和USP30直接去泛素化Parkin底物(包括TOM20、TOM70、VDACs等),抑制Parkin介导的线粒体自噬,但不会影响Parkin自身泛素化以及Parkin靶向线粒体的过程[35~37]。而USP8可直接对Parkin本身去泛素化,促进线粒体自噬的发生。Lys6(K6)位点连接的Ub以某种方式阻碍Parkin与线粒体Ub链结合,USP8通过选择性地移除连接在Parkin Lys6位上的Ub,使Parkin靶向至去极化线粒体,从而促进Parkin介导的线粒体自噬[38]。USPs和Parkin通过泛素化和去泛素化来达到线粒体上泛素链的稳态平衡。
3.2 PTEN-L磷酸酶通过去磷酸化Ub负调控线粒体自噬
研究表明,Ub的Ser65被PINK1磷酸化后,pSer65-Ub可影响Ub的结构和泛素链的组装,同时大部分去泛素化酶(包括USP8、USP15和USP30)不能对磷酸化的泛素链进行水解[39]。因此,Ub的去磷酸化是负调控线粒体自噬的关键。磷酸酶及张力蛋白同源物(phosphatase and tensin homolog,PTEN)是一种常见的肿瘤抑制因子,能够对磷脂酰肌醇三磷酸(phosphatidylinositol-3,4,5-trisphosphate,PIP3)进行去磷酸化以抑制蛋白激酶B(protein kinase B,AKT)的活性,从而影响肿瘤细胞的增殖和生长。PTEN-L(PTEN-Long,也被称为PTENα)是PTEN的一个亚型,其N端比PTEN多了173个氨基酸,具有潜在的分泌信号,不仅能够从细胞中分泌出来,还能再次进入其他细胞内抑制磷脂酰肌醇3-激酶(phosphatidylinositol 3-kinase,PI3K)信号。PTEN-L负调控线粒体解偶联物羰基氰化物间氯苯腙(carbonyl cyanide m-chlorophenyl hydrazine,CCCP)、寡霉素和抗霉素(Oligomycin and antimycin,OA)等处理的不同线粒体受损情况下诱导的线粒体自噬,同时抑制Parkin的线粒体定位过程。PTEN-L依赖其蛋白磷酸酶活性,通过增强泛素样(ubiquitin-like,UBL)结构域和锌指结构域RING1的互作,使Parkin保持封闭构象从而抑制Parkin E3泛素连接酶活性。PTEN-L以pSer65-Ub链为靶点去磷酸化泛素,进而破坏线粒体自噬中的前馈机制[40]。这些研究表明PTEN-L和PINK1对于维持线粒体稳态具有重要的意义。
此外,PINK1可以不依赖Parkin通过招募自噬受体(如NDP52和OPTINEURIN)来调控自噬[41,42],表明PINK1除正调控线粒体自噬外,可能还有其他维持线粒体稳态的功能。综上所述,哺乳动物细胞可能通过正调控机制和负调控机制共同来维持健康的线粒体群体(图2)。然而,更多的参与线粒体自噬的负调控机制有待进一步研究。

图2 线粒体自噬的正调控和负调控平衡Fig.2 The balance of positive and negative regulation of mitophagy.
4 异常的线粒体自噬与疾病
为了维持一个健康的线粒体群体,功能失调的线粒体要么融入健康线粒体网络,要么通过线粒体自噬降解。线粒体自噬是一个复杂的、涉及多种蛋白质和细胞器的通路,通过清除功能紊乱的线粒体可以降低细胞内ROS水平,避免细胞走向凋亡或坏死。异常的线粒体自噬会影响线粒体群体健康,破坏细胞正常的生理机能,引起一系列包括神经退行性疾病、心脏疾病和肿瘤等在内的疾病。
4.1 线粒体自噬障碍与疾病
神经元的正常新陈代谢需要线粒体提供大量能量,线粒体功能紊乱常常会导致神经元的病变,因此,很多神经退行性疾病与线粒体自噬功能障碍密切相关。研究发现,PINK1和Parkin的突变都会导致遗传型PD的发生[43,44]。二者的缺失会导致线粒体自噬不能正常进行,氧化应激增加,有毒物质大量积累导致中脑黑质线粒体的损伤,最终使得多巴胺能神经元死亡[45,46]。近来的研究表明,PINK1和Parkin可以通过清除受损的线粒体来预防炎症和神经变性,从而减轻PD的表型[47]。另一PD相关基因ATP13A2也参与维持健康的线粒体群体[48],ATP13A2突变使得线粒体功能受损[49],表明线粒体清除受损是帕金森症的一个重要致病因素。PD相关基因F-box结构域包含蛋白(the F-box domain containing proteins,Fbxo7)可以通过与PINK1和Parkin互作来调控线粒体自噬,共同参与线粒体稳态的维持[50]。这些PD相关基因的研究明确了线粒体自噬障碍与PD的关系。
AD是老年人中最常见的神经退行性疾病,也是一种与线粒体自噬障碍密切相关的神经退行性疾病。受AD影响的神经元在疾病发生的早期就出现线粒体功能失调和ROS水平升高,导致β-淀粉样蛋白(amyloid-β protein,Aβ)沉积和Tau蛋白过度磷酸化这2种AD的主要病理特征的出现。研究表明,AD中线粒体自噬功能障碍导致受损线粒体的积累,加剧了氧化损伤和能量缺失,引起Aβ和Tau积累致使突触功能障碍和认知缺陷,这些又进一步使得线粒体自噬水平显著下降[51]。PINK/Parkin介导的线粒体自噬在AD进程中发挥着调节作用,在小鼠AD模型中过表达PINK1或Parkin可加强受损线粒体的自噬清除,并延缓AD的神经退行性进程[52,53]。
4.2 过度的线粒体自噬与疾病
在生理条件下,线粒体自噬需维持在一定水平来参与线粒体质量控制,过度的线粒体自噬对细胞的存活是有害的,可能导致细胞死亡从而引发各种疾病。由于神经元对高产能的特殊需求,线粒体质量控制对神经元功能至关重要,适当的线粒体自噬能起到神经保护的作用,而过度的线粒体自噬则会使神经元死亡。PINK1功能缺陷可以导致氧化压的提高和依赖Drp1的分裂增多使得线粒体自噬水平升高,而这种线粒体自噬水平的增强会导致PD的发生[54]。
浦肯野细胞是位于小脑皮质的一类特殊神经元,浦肯野细胞变性(Purkinje cell degeneration,pcd)是人类和小鼠遗传性共济失调的共同特征。pcd小鼠中存在自噬的过度激活和线粒体自噬增强,表明过多或异常的线粒体自噬会导致pcd,最终神经元死亡[55]。BNIP3是Bcl-2蛋白家族成员,仅含BH-3结构域的促凋亡蛋白,它可以通过与LC3互作来诱导过度的线粒体自噬,导致脑缺血后的迟发性神经元死亡[56]。
心肌细胞同样对产能有高需求,因此,自噬水平的高低也能决定其利弊。当受到轻微的生理压力刺激时,可诱导线粒体自噬提高线粒体周转,起到保护作用。当心脏持续超负荷时,超生理水平的线粒体自噬会导致线粒体的过度清除,使得ATP生成受阻、心肌纤维化及病理性重构,最终导致心力衰竭[57]。
5 展望
线粒体自噬是把双刃剑,正常水平的线粒体自噬是机体应对受损线粒体的一种防御机制,可以起到保护神经元和心肌细胞等的作用;但是过度的线粒体自噬使得线粒体循环异常,能量代谢随之紊乱,最终导致细胞死亡。巨自噬相关蛋白的缺陷,以及与线粒体和溶酶体相关蛋白的缺陷都可能导致线粒体自噬的功能障碍。巨自噬缺陷会引起多重系统紊乱的人类疾病,如Vici综合征;溶酶体蛋白的丢失也与很多临床上表型较为严重的人类疾病相关,如Danon病、Pompe病及多种硫酸酯酶缺乏症等,表明自噬的完全缺陷会导致严重的细胞缺陷[58]。相对于巨自噬通路的障碍产生的严重表型,其亚通路线粒体自噬障碍并不会对机体造成如此大的危机。这说明线粒体自噬在线粒体整体维护中发挥相对较小的作用,或者有其他替代的线粒体自噬通路参与。但线粒体自噬障碍的现象却是在多种疾病中发现的,因此,更多关于线粒体自噬的机制亟待研究。
近年来,酵母和哺乳动物细胞中的线粒体自噬机制早已是研究热点,但不同因素或不同组织发生的线粒体自噬可能有不同的途径参与,更多的线粒体自噬机制的研究仍欠缺,有待进一步深入探究。功能障碍的线粒体会导致胞内ROS的累积,逐渐导致细胞死亡,调控线粒体自噬则可能逆转细胞的命运。因而,对线粒体自噬调控机制方面的深入研究,可为现阶段癌症、肿瘤、神经变性等疾病提供新的治疗方向和新的治疗靶标。
猜你喜欢
湖北农业科学(2022年11期)2022-07-18
波谱学杂志(2022年1期)2022-03-15
实用肿瘤学杂志(2020年4期)2020-12-08
医学综述(2020年11期)2020-02-16
天津医科大学学报(2019年6期)2019-08-13
分析化学(2017年12期)2017-12-25
天津科技大学学报(2016年1期)2016-02-28
安徽医科大学学报(2015年9期)2015-12-16
浙江大学学报(农业与生命科学版)(2015年4期)2015-12-15
中国医学科学院学报(2015年5期)2015-03-01
