
基于迁移学习的MHC-I型抗原表位呈递预测
2019-11-28 12:04胡伟澎李佑平张秀清
遗传 2019年11期
胡伟澎,李佑平,张秀清
基于迁移学习的MHC-I型抗原表位呈递预测
胡伟澎1,2,3,李佑平2,3,4,张秀清2,3,4
1. 华南理工大学生物科学与工程学院,广州 510006 2. 深圳华大生命科学研究院,深圳 518083 3. 华大吉诺因,武汉4300794 4. 中国科学院大学华大教育中心,深圳 518083
基于新抗原的肿瘤免疫治疗,抗原呈递的准确预测是筛选T细胞特异性表位的关键步骤。质谱鉴定的表位数据对建立抗原呈递预测模型具有重要价值。尽管近年来质谱数据的积累持续增加,但是大部分人类白细胞抗原(human leukocyte antigen, HLA)分型所对应的多肽数量相对较少,无法建立可靠的预测模型。为此,本研究尝试利用迁移学习的方法,先利用混合分型的表位数据建立模型以识别抗原表位的共同特征,在此预训练模型的基础上再利用分型特异性数据建立抗原呈递预测模型Pluto。在相同的验证集上,Pluto的平均0.1%阳性预测值(positive predictive value, PPV)比从头训练的模型高0.078。在外部的质谱数据独立评估上,Pluto的平均0.1% PPV为0.4255,高于从头训练模型(0.3824)和其他主流工具,包括MixMHCpred (0.3369)、NetMHCpan4.0-EL (0.4000)、NetMHCpan4.0-BA (0.3188)和MHCflurry (0.3002)。此外,在免疫原性预测评估上,Pluto相对于其他工具也能找到更多的新抗原。Pluto开源网址:https://github.com/weipenegHU/Pluto。
免疫治疗;新抗原;抗原呈递;深度学习;迁移学习
肿瘤细胞内含有肿瘤特异性突变位点的蛋白质能够被消化为不同长度的多肽,含有突变的多肽能够在内质网中与主要组织相同性复合体(major histocompatibility complex, MHC)结合形成多肽-MHC复合物然后被呈递到细胞表面,如果多肽-MHC复合物被T细胞特异性识别,即能够引起肿瘤细胞的凋亡,这种多肽被称为新抗原。近年来,基于新抗原的肿瘤免疫疗法在不同癌种的治疗中取得令人瞩目的突破[1~7],而且新抗原对于预测肿瘤疗效和病人预后具有重要价值[8~12]目前筛选新抗原的主流方法是通过亲和力预测工具预测多肽能否和人类白细胞抗原(human leukocyte antigen, HLA)结合,例如NetMHCpan系列工具[13~15]和MHCflurry[16]等。但是,这些工具使用的训练数据大部分来源于体外实验,不能真实反应细胞内多肽与HLA结合的情况。随着质谱技术的发展,科学家们能够直接获得呈递到细胞表面的多肽数据,相对于传统的经体外实验得到的亲和力数据,这些质谱数据更加真实地反应了多肽在细胞内加工到呈递的自然过程,包含更多的信息。随着质谱数据的积累以及质谱数据对多肽免疫原性预测的重要性得到越来越多的重视[17,18],基于质谱数据训练的抗原呈递预测模型也随之出现,例如MixMHCpred[19,20]和EDGE[17]。
虽然目前已经积累了一定数量的质谱鉴定的抗原表位数据,但对应到每个HLA分型的质谱数据并不均匀,大部分的HLA分型只有数千条多肽数据,有的更只有数百条。在这种情况下,并不能开发出可靠的分型特异性的抗原呈递预测模型。迁移学习或许能够帮助改善目前的这种状况,其基本原理是利用在一个相似任务上学习到的经验转移到最终需要解决的任务上,通常前者拥有大量的数据,而后者只有少量的数据。为了验证上述猜想,本研究先利用混合分型的MHC-I亚型抗原表位数据(是指训练数据由对应不同MHC-I亚型的抗原表位组成)来训练一个模型以区分抗原表位与普通的蛋白质多肽,再利用另外的包括16个HLA分型的单分型抗原表位数据在预训练模型的基础上训练最终的分型特异性抗原呈递模型,称之为Pluto。然后,在相同的验证集上评估了Pluto相对于从头训练模型的优势,并在独立验证集上比较了其与目前主流软件的表现。Pluto模型有望为相关工作提供新的思路以及对免疫治疗领域做出有益的贡献。
1 材料与方法
1.1 训练集构建
预训练模型用到的阳性集来源于Pearson等[21]和Bassani-Sternberg等[22]产生的数据以及SysteMHC质谱多肽数据库[23]。将这些数据集合并后,剔除长度小于8以及大于14的多肽,然后根据多肽和HLA分型去重,总共得到接近16万的多肽数据(表1)。阴性集来源于人类蛋白组的随机切割的多肽(剔除出现在阳性数据集中的多肽),从中挑取与阳性集等量的阴性多肽与阳性集合并构成训练集,然后从训练集中各挑取5000条阳性多肽和5000条阴性多肽构成验证集。
抗原呈递模型中用到的阳性训练数据来源于Abelin等[24]研究的16个单分型细胞系,包括A01:01、A02:01、A02:03、A02:04、A02:07、 A03:01、 A24:02、A29:02、A31:01、A68:02、B35:01、B44:02、B44:03、B51:01、B54:01和B57:01,总共约有2.7万条的多肽(表1),分别为这16个分型单独建模。每个分型的数据按照8:2的比例划分为阳性训练集和阳性验证集。从随机切割的蛋白质多肽中挑取阳性训练集数据100倍的阴性多肽与阳性训练集合并构成训练集,挑取阳性验证集数据999倍的阴性多肽与阳性验证集合并构成验证集。
本研究构建的模型主要对长度在8~14的短肽进行预测,因此预训练模型和抗原呈递模型使用的多肽先利用通配符’X’把多肽的长度统一为14肽,然后利用热编码将每条多肽编码为294维(14×21,算上通配符‘X’,每个氨基酸需要编码为21维向量)的向量。
1.2 模型训练
预训练的模型由输入层、5层隐藏层和输出层组成(图1A),其中5层隐藏层包含的神经元数目分别为100、30、100、30和10,第一个和第三个隐藏层采用dropout(dropout rate=0.4)来控制模型的过拟合,各隐藏层均使用exponential linear unit (ELU)作为激活函数。本研究采用批次梯度下降的方法训练预训练模型,每个批次包含1024条多肽(阳性和阴性多肽各一半),总共迭代100次。采用5层交叉验证的方法来评估预训练模型的准确率。
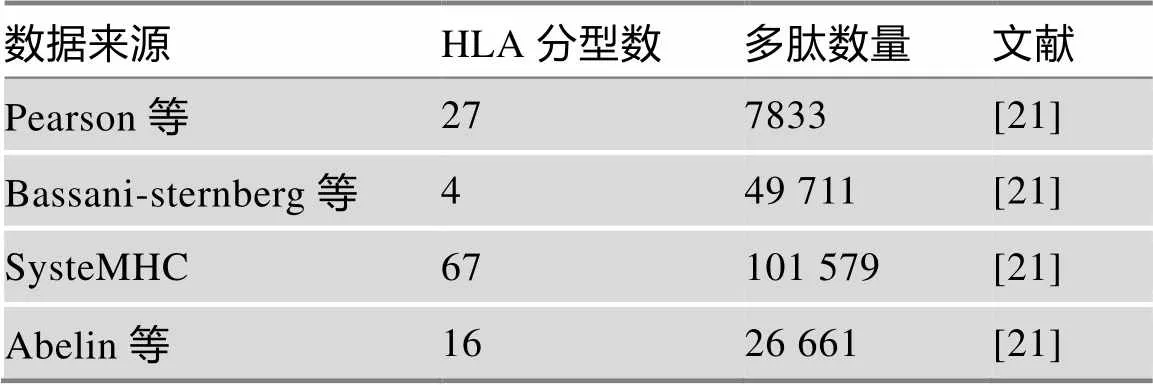
表1 训练集总结
Pluto的结构是在预训练模型结构的基础上,在最后一层隐藏层和输出层之间增加了一个隐藏层,这层隐藏层之前的神经元参数均使用预训练模型中对应神经元的参数,而且不再对这些神经元进行训练,而只对新增的隐藏层和输出层的神经元训练。同样采用批次梯度下降的方法训练模型,每个批次包含全部的阳性多肽以及10倍的阴性多肽,总共迭代1000次。每迭代一次,利用训练好的模型对验证集进行预测打分,根据分数大小进行排序并统计排名前0.1%的结果(阳性多肽的数目)中的阳性预测值(positive predictive value, PPV)。最后根据0.1%PPV的表现选择最终的模型。采用0.1%PPV评估标准是因为根据之前报道[25~27],细胞内能被呈递的多肽若占整个人类蛋白组的0.1%,因此该评估标准更能够反映实际情况。模型的实现和训练均采用Tensorflow框架[28]。

图1 Pluto的构建过程
A:预训练模型的结构;B: Pluto的结构中,前5层隐藏层使用的权重来源于预训练模型的结构,训练过程中对这些迁移过来的权重锁定,即这些权重在训练过程中不会改变,并且只对新增加的隐藏层和输出层的权重进行训练。
1.3 外部质谱数据评估
收集了Trolle等[29]产生的HeLa单分型细胞系多肽数据对模型进行独立评估。把这些质谱多肽与999倍的阴性多肽合并构建成测试集,生成的测试集用于评估Pluto、从头训练模型以及MixMHCpred、NetMHCpan4.0-EL、NetMHCpan4.0-BA和MHCflurry的0.1% PPV。
1.4 免疫原性评估
收集了Stronen等[30]和Gros等[31]经实验验证具有免疫原性的多肽。因为Stronen等是利用四聚体实验直接对包含突变的多肽进行验证的,所以每条多肽是否具有免疫原性是明确的。而Gros等是利用串联迷你基因(tandem mini-gene, TMG)验证的,把这些TMG切成长度为8~11个氨基酸,包含突变位点的重叠连续多肽。来自于没有免疫原性的TMG的多肽被标记为阴性数据。来自于具有免疫原性的TMG但是没有经过多肽负载实验验证的多肽会被剔除,因为不能确定这些多肽能否被T细胞识别,其他多肽则按照多肽负载实验验证的结果标记为阳性和阴性多肽。然后利用Pluto、MixMHCpred、NetMHCpan4.0-EL、NetMHCpan4.0-BA和MHCflurry对这些多肽进行预测,并比较这些工具对免疫原性多肽的排位。
2 结果与分析
2.1 预训练能提高抗原呈递预测模型的表现
假设预训练模型从大量呈递的抗原表位中学习到一些非分型特异性的特征,并且能够提高分型特异性抗原呈递预测模型的表现。为验证此假设,本研究利用单分型训练集从头训练Pluto整个网络的全部参数,而不利用预训练模型训练好的参数,并且在相同的验证集上和Pluto的表现作比较(图2A)。
通过分析,经过混合分型的表位数据训练的预训练模型五层交叉验证的平均准确率为90.77%,说明模型学习到一些能够将抗原表位与普通蛋白质多肽区分开来的特征。接下来在16个单分型验证集上评估Pluto与从头训练模型的0.1%PPV,结果发现Pluto的0.1%PPV在所有验证集上都比没有经预训练的模型要高,平均0.1% PPV提升了0.078。然后观察了训练集大小与模型表现提升之间的关系,从图2B中可以发现这样一种趋势:迁移学习对数据量小的分型的表现提升帮助更加明显,而对数据量较大的分型来说,迁移学习对模型的提升则比较小。
因此,上述结果表明预训练模型能够学习到不同分型抗原表位的共同特征,并且能够帮助提高分型特异性的抗原呈递预测模型的表现,而提升的幅度可能受到抗原呈递预测模型的训练集大小的影响。
2.2 在质谱数据上独立评估模型表现
利用Trolle等[29]产生的单分型质谱数据对Pluto的使用效果进行评估,并与从头训练的模型和主流预测工具作(包括MixMHCpred (v2.0)[19,20]、NetMHCpan4.0-EL[13]、NetMHCpan4.0-BA[13]和MHCflurry[16])进行比较,结果发现 Pluto在独立测试集上的平均0.1% PPV为0.4255,显著优于从头训练模型、MixMHCpred、NetMHCpan4.0-BA和MHCflurry,这些模型的平均0.1% PPV分别为0.3824、0.3369、0.3188、0.3002 (= 0.02538、0.002035、0.01102、0.01929,paired-test)。值得注意的是,虽然Pluto的平均0.1% PPV没有显著高于NetMHCpan4.0-EL (0.42550.4000,= 0.05311),但是在每个分型上Pluto的表现都要好于NetMHCpan4.0-EL (图3)。
MixMHCpred是基于位置特异性打分矩阵(position specific scoring matrix, PSSM)以及只用质谱数据训练的抗原呈递预测模型。PSSM属于线性模型的一种,它基于的假设是多肽的每个位置都是独立,而从图3的分析结果看,从头训练模型和NetMHCpan4.0-EL表现要显著优于MixMHCpred (= 0.03048, 5.674e-05, paired-test),因此推测多肽的不同位置之间可能存在一定的联系,而不是单纯的线性关系(本研究选择从头训练模型与NetMHCpan4.0- EL和MixMHCpred比较,是因为它们都是抗原呈递预测模型,而NetMHCpan4.0-BA和MHCflurry是亲和力预测模型)。

图2 Pluto与从头训练模型的性能比较
A:在16个单分型相同的验证集上Pluto的0.1% PPV表现都要优于从头训练的模型;B:预训练模型对Pluto表现提升的幅度受训练集大小的影响。

图3 在外部质谱数据上进行独立评估
Pluto的平均0.1% PPV要显著高于从头训练模型,MixMHCpred,NetMHCpan4.0-BA和MHCflurry。Pluto的平均0.1%PPV虽然没有显著高于NetMHCpan4.0-EL,但是在每个分型上的表现都要高于NetMHCpan4.0-EL。*代表<0.05,**代表<0.005 (paired-test)。
综上所述,通过独立评估,本研究验证了Pluto能够达到甚至优于目前主流的抗原呈递预测工具的水平。
2.3 肿瘤新抗原鉴定
为评估Pluto预测抗原呈递的能力能否用于寻找新抗原,本研究从Stronen等[30]和Gros等[31]的研究中收集了7条经实验验证具有免疫原性的多肽,并利用这些多肽评估Pluto、MixMHCpred、NetMHCpan4.0-EL、NetMHCpan4.0-BA和MHCflurry预测新抗原的能力。结果如表2所示,在每个病人排名前10的多肽中,Pluto能够找回7条免疫原性多肽中的4条,MixMHCpred和NetMHCpan4.0-EL能够找回其中的两条,而NetMHCpan4.0-BA和MHCflurry只能找到其中的1条。因此,评估结果证明了Pluto对于鉴定肿瘤新抗原具有重要价值。
3 讨论
抗原呈递的准确预测是判断新抗原能否激活新抗原特异性T细胞从而杀死肿瘤细胞的关键一步。虽然近几年来质谱技术飞速发展,积累了不少通过质谱鉴定的抗原表位数据,但是对于特定分型来说,每个分型对应的抗原表位数据还不是很多,对于建立一个基于深度学习的分型特异性的抗原表位预测模型来说是不足够的。因此本研究利用迁移学习的方法,从大量的混合分型抗原表位数据和蛋白质组中随机多肽数据建立了一个深度学习模型以识别抗原表位是否存在一些共性,使之能够与普通的多肽区分开。然后在预训练模型的基础上,利用分型特异性的数据训练了抗原呈递预测模型Pluto。
本研究首先展示了预训练模型能够将大部分的抗原表位与蛋白质组的普通多肽分开,说明模型学到了抗原表位的一些共同特征。但是因为深度学习本身的原因,预训练模型学习到哪些共同特征尚无法明确,值得后续研究给予重点关注。Pluto的表现相对于从头训练的模型的表现有明显的提升,但是提升的幅度受到分型特异性的训练集大小的影响。分析造成这种影响的原因可能有3个:一是随着分型特异性的抗原表位数据增加,所包含的信息量更多,与混合分型的抗原表位提供的信息有更大重合,这导致预训练模型学习到的特征起到的作用更小;二是模型可能已经接近饱和状态,增加数据量对模型提高帮助不大;三是随着数据量的增加,需要建立更加复杂的网络以学习更多的特征才能提高模型的表现。在利用外部数据进行独立评估以及鉴定新抗原上,Pluto的表现也优于从头训练的模型以及这个领域的其他主流工具。本文中用到的所有训练数据和评估数据都可以从https://github.com/weipenegHU/Pluto获取。
抗原表位需要经过源蛋白的表达,源蛋白经蛋白酶体消化切割后产生的多肽被转运到内质网内部与MHC-I分子结合,最后才能被呈递到细胞表面。在本研究中,Pluto只是根据抗原表位序列自身包含的信息来判定多肽能否被呈递到细胞表面,而序列本身提供的信息是非常有限的。据文献报道,多肽的表达量对抗原呈递具有很大的影响[17,24,32]。此外,抗原表位的上下游序列能够帮助预测多肽能否被蛋白酶体切割[17,24,33]。还有文献报道能够产生抗原表位的蛋白质只占细胞内所有蛋白质的一部分[21],以及蛋白质中存在产生抗原表位的热点[34]。相信这些特征能够进一步提高Pluto的表现,开发和利用这些特征将是未来工作的重要方向。

表2 Pluto与主流工具对免疫原性多肽的排名
虽然根据抗原呈递的可能性挑选新抗原具有一定效果[17],但是被呈递的多肽不一定具有免疫原性(多肽的免疫原性是指多肽能否被T细胞识别从而杀死肿瘤细胞)[35~37]。所以除了抗原呈递预测外,对多肽的免疫原性预测也具有重要意义。但是目前因为免疫原性数据缺乏积累,所以难以建立多肽免疫原性预测模型。未来通过共同协作产生更多的免疫原性数据,更好的实验方法来了解TCR和多肽-MHC分子的相互作用[38,39]以产生更大的数据集和对免疫原性更深的生物学认识,最终能够更准确地预测免疫原性。
综上所述,本研究利用迁移学习的方法建立了一个新的抗原呈递预测工具Pluto,其表现显著优于目前主流的预测软件。同时,这些结果说明了迁移学习对解决目前因分型特异性的抗原表位数据不足而难以建立一个可靠的抗原呈递预测模型的问题有所帮助。
[1] Gros A, Parkhurst MR, Tran E, Pasetto A, Robbins PF, Ilyas S, Prickett TD, Gartner JJ, Crystal JS, Roberts IM, Trebska-Mcgowan K, Wunderlich JR, Yang JC, Rosenberg SA. Prospective identification of neoantigen-specific lymphocytes in the peripheral blood of melanoma patients., 2016, 22(4): 433–438.
[2] Malekzadeh P, Pasetto A, Robbins PF, Parkhurst MR, Paria BC, Jia L, Gartner JJ, Hill V, Yu Z, Restifo NP, Sachs A, Tran E, Lo W, Somerville RPT, Rosenberg SA, Deniger DC. Neoantigen screening identifies broad TP53 mutant immunogenicity in patients with epithelial cancers., 2019, 129(3): 1109–1114.
[3] Robbins PF, Lu YC, El-Gamil M, Li YF, Gross C, Gartner J, Lin JC, Teer JK, Cliften P, Tycksen E, Samuels Y, Rosenberg SA. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells., 2013, 19(6): 747–752.
[4] Sahin U, Derhovanessian E, Miller M, Kloke BP, Simon P, Löwer M, Bukur V, Tadmor AD, Luxemburger U, Schrörs B, Omokoko T, Vormehr M, Albrecht C, Paruzynski A, Kuhn AN, Buck J, Heesch S, Schreeb KH, Müller F, Ortseifer I, Vogler I, Godehardt E, Attig S, Rae R, Breitkreuz A, Tolliver C, Suchan M, Martic G, Hohberger A, Sorn P, Diekmann J, Ciesla J, Waksmann O, Brück A K, Witt M, Zillgen M, Rothermel A, Kasemann B, Langer D, Bolte S, Diken M, Kreiter S, Nemecek R, Gebhardt C, Grabbe S, Höller C, Utikal J, Huber C, Loquai C, Türeci O. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer., 2017, 547(7662): 222–226.
[5] Tran E, Ahmadzadeh M, Lu YC, Gros A, Turcotte S, Robbins PF, Gartner JJ, Zheng Z, Li YF, Ray S, Wunderlich JR, Somerville RP, Rosenberg SA. Immunogenicity of somatic mutations in human gastrointestinal cancers., 2015, 350(6266): 1387–1390.
[6] Tran E, Robbins PF, Lu YC, Prickett TD, Gartner JJ, Jia L, Pasetto A, Zheng Z, Ray S, Groh EM, Kriley IR, Rosenberg SA. T-Cell transfer therapy targeting mutant KRAS in cancer., 2016, 375(23): 2255–2262.
[7] Zacharakis N, Chinnasamy H, Black M, Xu H, Lu YC, Zheng Z, Pasetto A, Langhan M, Shelton T, Prickett T, Gartner J, Jia L, Trebska-Mcgowan K, Somerville RP, Robbins PF, Rosenberg SA, Goff SL, Feldman SA. Immune recognition of somatic mutations leading to complete durable regression in metastatic breast cancer., 2018, 24(6): 724–730.
[8] Strickland KC, Howitt BE, Shukla SA, Rodig S, Ritterhouse LL, Liu JF, Garber JE, Chowdhury D, Wu CJ, D'andrea AD. Association and prognostic significance of BRCA1/2-mutation status with neoantigen load, number of tumor-infiltrating lymphocytes and expression of PD-1/ PD-L1 in high grade serous ovarian cancer., 2016, 7(12): 13587-13598.
[9] Lu HZ,Wang DK,Wang Z. Correlation analysis of the prognosis of HPV positive oropharyngeal cancer patients with T cell infiltration and neoantigen load., 2019, 41(8): 725–735.卢涣滋, 王迪侃, 王智. HPV阳性口咽癌患者预后与T细胞浸润和新抗原负荷相关性分析. 遗传, 2019, 41(8): 725–735.
[10] Brown SD, Warren RL, Gibb EA, Martin SD, Spinelli JJ, Nelson BH, Holt RA. Neo-antigens predicted by tumor genome meta-analysis correlate with increased patient survival., 2014, 24(5): 743–750.
[11] Shukla SA, Howitt BE, Wu CJ, Konstantinopoulos PA. Predicted neoantigen load in non-hypermutated endometrial cancers: Correlation with outcome and tumor-specific genomic alterations., 2016, 19: 42–45.
[12] Sa HL, Ma KW, Gao Y, Wang DQ. Predictive value of tumor mutation burden in immunotherapy for lung cancer., 2019, 22(6): 380–384.撒焕兰, 马克威, 高勇, 王德强. 肿瘤突变负荷对肺癌免疫治疗疗效的预测价值. 中国肺癌杂志, 2019, 22(6): 380–384.
[13] Jurtz V, Paul S, Andreatta M, Marcatili P, Peters B, Nielsen M. NetMHCpan-4.0: Improved peptide-MHC class I interaction predictions integrating eluted ligand and peptide binding affinity data., 2017, 199(9): 3360–3368.
[14] Nielsen M, Andreatta M. NetMHCpan-3.0; improved prediction of binding to MHC class I molecules integrating information from multiple receptor and peptide length datasets., 2016, 8(1): 33.
[15] Nielsen M, Lundegaard C, Blicher T, Lamberth K, Harndahl M, Justesen S, Røder G, Peters B, Sette A, Lund O, Buus S. NetMHCpan, a method for quantitative predictions of peptide binding to any HLA-A and -B locus protein of known sequence., 2007, 2(8): e796.
[16] O'donnelL TJ, Rubinsteyn A, Bonsack M, Riemer AB, Laserson U, Hammerbacher J. MHCflurry: open-source class I MHC binding affinity prediction., 2018, 7(1): 129–132 e4.
[17] Bulik-Sullivan B, Busby J, Palmer CD, Davis MJ, Murphy T, Clark A, Busby M, Duke F, Yang A, Young L, Ojo NC, Caldwell K, Abhyankar J, Boucher T, Hart MG, Makarov V, Montpreville VT, Mercier O, Chan TA, Scagliotti G, Bironzo P, Novello S, Karachaliou N, Rosell R, Anderson I, Gabrail N, Hrom J, Limvarapuss C, Choquette K, Spira A, Rousseau R, Voong C, Rizvi NA, Fadel E, Frattini M, Jooss K, Skoberne M, Francis J, Yelensky R. Deep learning using tumor HLA peptide mass spectrometry datasets improves neoantigen identification., 2018, 37(1): 55–63
[18] Gfeller D, Bassani-Sternberg M. Predicting antigen presentation—what could we learn from a million peptides?, 2018, 9: 1716.
[19] Bassani-Sternberg M, Chong C, Guillaume P, Solleder M, Pak H, Gannon PO, Kandalaft LE, Coukos G, Gfeller D. Deciphering HLA-I motifs across HLA peptidomes improves neo-antigen predictions and identifies allostery regulating HLA specificity., 2017, 13(8): e1005725.
[20] Gfeller D, Guillaume P, Michaux J, Pak HS, Daniel RT, Racle J, Coukos G and Bassani-Sternberg M. The length distribution and multiple specificity of naturally presented HLA-I ligands., 2018, 201(12): 3705–3716.
[21] Pearson H, Daouda T, Granados DP, Durette C, Bonneil E, Courcelles M, Rodenbrock A, Laverdure JP, Coté C, Mader S, Lemieux S, Thibault P, Perreault C. MHC class I-associated peptides derive from selective regions of the human genome., 2016, 126(12): 4690–4701.
[22] Bassani-Sternberg M, Pletscher-Frankild S, Jensen LJ, Mann M. Mass spectrometry of human leukocyte antigen class I peptidomes reveals strong effects of protein abundance and turnover on antigen presentation., 2015, 14(3): 658–673.
[23] Shao W, Pedrioli PGA, Wolski W, Scurtescu C, Schmid E, Vizcaíno JA, Courcelles M, Schuster H, Kowalewski D, Marino F, Arlehamn CSL, Vaughan K, Peters B, Sette A, Ottenhoff THM, Meijgaarden KE, Nieuwenhuizen N, Kaufmann SHE, Schlapbach R, Castle JC, Nesvizhskii A I, Nielsen M, Deutsch E W, Campbell D S, Moritz R L, Zubarev R A, Ytterberg A J, Purcell A W, Marcilla M, Paradela A, Wang Q, Costello CE, Ternette N, van Veelen PA, van Els CACM, Heck AJR, de Souza GA, Sollid LM, Admon A, Stevanovic S, Rammensee HG, Thibault P, Perreault C, Bassani-Sternberg M, Aebersold R, Caron E. The SysteMHC atlas project., 2018, 46(D1): D1237–D1247.
[24] Abelin JG, Keskin DB, Sarkizova S, Hartigan CR, Zhang W, Sidney J, Stevens J, Lane W, Zhang GL, Eisenhaure TM, Clauser KR, Hacohen N, Rooney MS, Carr SA, Wu CJ. Mass spectrometry profiling of HLA-Associated peptidomes in Mono-allelic cells enables more accurate epitope prediction., 2017, 46(2): 315–326.
[25] Vita R, Overton JA, Greenbaum JA, Ponomarenko J, Clark JD, Cantrell JR, Wheeler DK, Gabbard JL, Hix D, Sette A, Peters B. The immune epitope database (IEDB) 3.0., 2015, 43(Database issue): D405–412.
[26] Rammensee HG, Friede T, Stevanoviíc S. MHC ligands and peptide motifs: first listing., 1995, 41(4): 178–228.
[27] Hunt DF, Henderson RA, Shabanowitz J, Sakaguchi K, Michel H, Sevilir N, Cox AL, Appella E, Engelhard VH. Characterization of peptides bound to the class I MHC molecule HLA-A2.1 by mass spectrometry., 1992, 255(5049): 1261–1263.
[28] Abadi M, Agarwal A, Barham P, Brevdo E, Chen Z, Citro C, Corrado GS, Davis A, Dean J, Devin M, Ghemawat S, Goodfellow I, Harp A, Irving G, Isard M, Jia YQ, Jozefowicz R, Kaiser L, Kudlur M, Levenberg J, Mane D, Monga R, Moore S, Murray D, Olah C, Schuster M, Shlens J, Steiner B, Sutskever I, Talwar K, Tucker P, Vanhoucke V, Vasudevan V, Viegas F, Vinyals O, Warden P, Wattenberg M, Wicke M, Yu Y, Zheng XQ. Tensorflow: Large-scale machine learning on heterogeneous distributed systems.:1603.04467, 2016,
[29] Trolle T, Mcmurtrey CP, Sidney J, Bardet W, Osborn SC, Kaever T, Sette A, Hildebrand WH, Nielsen M, Peters B. The length distribution of class I-restricted T cell epitopes is determined by both peptide supply and MHC allele- specific binding preference., 2016, 196(4): 1480–1487.
[30] Strønen E, Toebes M, Kelderman S, van Buuren MM, Yang W, van Rooij N, Donia M, Böschen ML, Lund- Johansen F, Olweus J, Schumacher TN. Targeting of cancer neoantigens with donor-derived T cell receptor repertoires., 2016, 352(6291): 1337–1341.
[31] Gros A, Parkhurst MR, Tran E, Pasetto A, Robbins PF, Ilyas S, Prickett TD, Gartner JJ, Crystal JS, Roberts IM. Prospective identification of neoantigen-specific lymphocytes in the peripheral blood of melanoma patients., 2016, 22(4): 433–438.
[32] Hu WP, Qiu S, Li YP, Lin XX, Zhang L, Xiang HT, Han X, Chen L, Li S, Li WH, Ren Z, Hou GX, Lin ZL, Lu JL, Liu G, Li B, Lee LJ. EPIC: MHC-I epitope prediction integrating mass spectrometry derived motifs and tissue- specific expression profiles., 2019, 567081.
[33] Nielsen M, Lundegaard C, Lund O, Kesmir C. The role of the proteasome in generating cytotoxic T-cell epitopes: insights obtained from improved predictions of proteasomal cleavage., 2005, 57(1–2): 33–41.
[34] Müller M, Gfeller D, Coukos G, Bassani-Sternberg M. 'Hotspots' of antigen presentation revealed by human leukocyte antigen ligandomics for neoantigen prioritization., 2017, 8: 1367.
[35] Mcgranahan N, Furness AJ, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, Jamal-Hanjani M, Wilson GA, Birkbak NJ, Hiley CT, Watkins TB, Shafi S, Murugaesu N, Mitter R, Akarca AU, Linares J, Marafioti T, Henry JY, Van Allen EM, Miao D, Schilling B, Schadendorf D, Garraway LA, Makarov V, Rizvi NA, Snyder A, Hellmann MD, Merghoub T, Wolchok JD, Shukla SA, Wu CJ, Peggs KS, Chan TA, Hadrup SR, Quezada SA, Swanton C. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade., 2016, 351(6280): 1463–1469.
[36] Calis JJ, Maybeno M, Greenbaum JA, Weiskopf D, de Silva AD, Sette A, Keşmir C, Peters B. Properties of MHC class I presented peptides that enhance immunogenicity., 2013, 9(10): e1003266.
[37] Assarsson E, Sidney J, Oseroff C, Pasquetto V, Bui HH, Frahm N, Brander C, Peters B, Grey H, Sette A. A quantitative analysis of the variables affecting the repertoire of T cell specificities recognized after vaccinia virus infection., 2007, 178(12): 7890–7901.
[38] Bentzen AK, Such L, Jensen KK, Marquard AM, Jessen LE, Miller NJ, Church CD, Lyngaa R, Koelle DM, Becker JC, Linnemann C, Schumacher TNM, Marcatili P, Nghiem P, Nielsen M, Hadrup SR. T cell receptor fingerprinting enables in-depth characterization of the interactions governing recognition of peptide–MHC complexes., 2018, 36(12): 1191–11996.
[39] Bentzen AK, Marquard AM, Lyngaa R, Saini SK, Ramskov S, Donia M, Such L, Furness AJ, Mcgranahan N, Rosenthal R, Straten PT, Szallasi Z, Svane IM, Swanton C, Quezada SA, Jakobsen SN, Eklund AC, Hadrup SR. Large-scale detection of antigen-specific T cells using peptide-MHC-I multimers labeled with DNA barcodes., 2016, 34(10): 1037–1045.
MHC-I epitope presentation prediction based on transfer learning
Weipeng Hu1,2,3, Youping Li2,3,4, Xiuqing Zhang2,3,4
Accurate epitope presentation prediction is a key procedure in tumour immunotherapies based on neoantigen for targeting T cell specific epitopes. Epitopes identified by mass spectrometry (MS) is valuable to train an epitope presentation prediction model. In spite of the accelerating accumulation of MS data, the number of epitopes that match most of human leukocyte antigens (HLAs) is relatively small, which makes it difficult to build a reliable prediction model. Therefore, this research attempted to use the transfer learning method to train a model to learn common features among the mixed allele specific epitopes. Then based on this pre-trained model, we used the allele-specific epitopes to train the final epitope presentation prediction model, termed Pluto. The average 0.1% positive predictive value (PPV) of Pluto outperformed the prediction model without pretraining with a margin of 0.078 on the same validation dataset. When evaluating Pluto on external HLA eluted ligand datasets, Pluto achieved an averaged 0.1% PPV of 0.4255, which is better than the prediction model without pretraining (0.3824) and other popular methods, including MixMHCpred (0.3369), NetMHCpan4.0-EL (0.4000), NetMHCpan4.0-BA (0.3188) and MHCflurry (0.3002). Moreover, when it comes to the evaluation of predicting immunogenicity, Pluto can identify more neoantigens than other tools. Pluto is publicly available at https://github.com/weipenegHU/Pluto.
immunotherapy; neoantigen; epitope presentation; deep learning; transfer learning
2019-06-21;
2019-09-17
国家自然科学基金项目(编号:81702826,81772910),深圳市科创委项目(编号:JCYJ20170303151334808)和深圳市经信委项目(编号:20170731162715261)资助[Supported by the National Natural Science Foundation of China (Nos. 81702826, 81772910 ), Science, Technology and Innovation Commission of Shenzhen Municipality (No. JCYJ20170303151334808) and Shenzhen Municipal Government of China (No. 20170731162715261)]
胡伟澎,硕士研究生,专业方向:基因组学。E-mail: huweipeng@genomics.cn 李佑平,硕士研究生,专业方向:基因组学。E-mail: liyouping@genomics.cn 胡伟澎和李佑平并列第一作者。
张秀清,博士,教授,研究方向:基因组学及免疫治疗。E-mail: zhangxq@genomics.cn
10.16288/j.yczz.19-155
2019/11/8 13:27:56
URI: http://kns.cnki.net/kcms/detail/11.1913.R.20191107.1628.005.html
(责任编委: 赵要凤)
猜你喜欢
温州医科大学学报(2019年4期)2019-04-28
现代园艺(2017年13期)2018-01-19
上海农业学报(2017年4期)2017-04-10
中国免疫学杂志(2017年1期)2017-01-17
现代检验医学杂志(2016年3期)2016-11-15
特产研究(2016年3期)2016-04-12
中国学术期刊文摘(2016年1期)2016-02-13
畜牧兽医学报(2015年3期)2015-07-05
医学研究杂志(2015年6期)2015-07-01
药学与临床研究(2015年4期)2015-06-05
