
肾病综合征患者肠道菌群宏基因组学
2019-12-20 07:46刘永平郑学明王茹冰解冰孔德华吴晓蓉傅行礼
中国老年学杂志 2019年24期
刘永平 郑学明 王茹冰 解冰 孔德华 吴晓蓉 傅行礼
(镇江市丹徒区中医院,江苏 镇江 212028;2江苏大学医学院;3南京市高淳人民医院)
人体是一个非常复杂的生态系统,在我们的体表、肠道和口腔中活跃着非常丰富的微生物〔1,2〕。其中,人类肠道微生物最为丰富,总数在1 012~1 014之间,是人体细胞数量的10倍〔3〕。 成年人胃肠道微生物的其中绝大部分是细菌,主要类群为拟杆菌门和硬壁菌门,而放线菌门、变形菌门和疣微菌门数量次之〔4〕。越来越多的研究表明肠道微生物与人体的健康密切相关,肠道菌群的失调可以导致自身免疫性疾病、过敏反应、肥胖、炎症性肠(IBD)、糖尿病等疾病〔5~7〕。由于绝大部分的肠道微生物无法通过传统的分离培养来鉴定,目前的研究手段主要是肠道宏基因组测序〔8〕。宏基因组学研究借助于大规模测序和生物信息学分析,能够发现大量过去无法得到的未知微生物新基因或新的基因簇,这对于了解胃肠道微生物的群落组成、进化历程和代谢特点,挖掘具有应用潜力的新基因具有重要意义。肾脏疾病的发生和肠道微生物有密切关系。Zheng等〔9〕发现克雷伯氏菌能促进三聚氰胺对肾脏的毒性,表明患者肾衰竭与其肠道菌群的组成和代谢活动密切相关。美国学者Vaziri等〔10〕研究了尿毒症或饮食和药物干预慢性肾脏病对肠道微生物的改变,研究人员通过研究粪便中提取的肠道微生物的宏基因组发现终末期肾脏病患者和健康人两组之间有 190种细菌的分类有显著差异:短状杆菌科、肉杆菌科、肠杆菌科、盐单胞菌科等细菌在终末期肾脏病(ESRD)患者中均显著增加。Anders等〔11〕提出了肠道菌群对慢性肾病(CKD)和ESRD患者有潜在的免疫调节作用并讨论了尿毒症是如何引起患者肠道微生物组失调。肾病综合征(NS)可由多种病因引起,最基本的特征是大量蛋白尿、低蛋白血症、水肿和高脂血症及其他代谢紊乱为特征的一组临床症候群。目前对NS患者肠道微生物宏基因组还缺少研究。本研究主要目的是应用肠道微生物宏基因组学的方法来研究NS患者与健康人群肠道菌群在物种多样性、菌落结构上的差异。
1 材料与方法
1.1样品的准备和DNA的抽提 从镇江市丹徒区中医院收集正常人和NS患者的粪便标本分别为8 份和30份。粪便标本储存在含有DNA稳定剂的粪便收集器中(Invitek,Germany),保存在-80℃冰箱备用。样品中微生物宏基因组DNA的提取选择德国Invitek公司的试剂盒PSP®Spin Stool DNA Plus Kit。提取的基因组DNA后需要经过质检(1.0%琼脂糖凝胶电泳及紫外分光光度计测OD260/OD280)后可进行下一步的 PCR 扩增实验。
1.2PCR扩增和产物的回收 设计细菌16S rDNA基因V3~V5区的通用引物,进行PCR扩增。正向引物338F:ACTCCTACGGGAGGCAGCAG,反向引物806R:GGACTACHVGGGTWTCTAAT (注:H:A/T/C,V:G/A/C,W:A/T)。为保证后续数据分析的准确性及可靠性,我们选择高保真的DNA聚合酶TransStart FastPfu DNA Polymerase(全式金生物)进行PCR反应并且①尽可能使用低循环数扩增;②保证每个样本扩增的循环数一致。首先随机选取具有代表性的样本进行预实验确定能够扩增出浓度合适的产物的最低循环数,再用这一循环数扩增所有样品中的16S rDNA基因V3~V5区。每个样本做3个重复,反应结束后将同一样本的PCR产物混合,用2%琼脂糖凝胶电泳检测,使用AxyPrepDNA凝胶回收试剂盒(AXYGEN公司)切胶回收PCR产物,Tris HCl洗脱,2%琼脂糖电泳检测。
1.3测序和质控 参照电泳初步定量结果,将PCR产物用QuantiFluorTM-ST蓝色荧光定量系统(Promega公司)进行定量,再按照每个样本的测序量要求进行相应比例的混合。用TruSeqTM DNA样品准备试剂盒进行Miseq文库构建,Illumina Miseq测序仪器上进行双端(PE)高通量测序。根据PE测序读长(reads)之间的重叠关系,将成对的reads拼接成一条序列,同时对reads的质量和合并的效果进行质控过滤,得到优化数据进行下一步的生物信息学分析。质控参数:①过滤reads尾部质量值20以下的碱基,设置50 bp的窗口,如果窗口内的平均质量值低于20 bp,从窗口开始截去后端碱基,过滤质控后50 bp以下的reads,去除含N碱基的reads;②根据PE reads之间的重叠关系,将成对reads拼接成一条序列,最小重叠长度为10 bp;③拼接序列的重叠区允许的最大错配比率为0.2,筛选不符合序列;④根据序列首尾两端的条形码和引物区分样品,并调整序列方向,条形码允许的错配数为0,最大引物错配数为2。
1.4生物信息学分析 用Usearch软件(vsesion 7.0 http://drive5.com/uparse/)对所有序列进行优化序列进行OTU(Operational Taxonomic Units)划分〔12〕。OTU分析步骤如下:①提取非重复序列;②去除没有重复的单序列;③按照97%相似性对非重复序列(不含单序列)进行OTU聚类,在聚类过程中去除嵌合体,得到OTU的代表序列。为了得到每个OTU对应的物种分类信息,采用Silva(版本128,http://www.arb-silva.de)对97%相似水平的OTU代表序列进行分类学分析,并分别在各个分类学水平上统计各样本的群落组成〔13〕。基于OTU的注释数据,我们得到每个样品中物种组成、群落结构等信息。进一步,在I-Sanger生信云(http://www.i-sanger.com/)上统计分析肾病综合征患者和正常人肠道细菌多样性,物种组成的差异。
2 结 果
2.1PCR结果 16S rDNA的扩增由于16S rDNA较长(1.5 kb),我们只能对其中经常变化的区域也就是可变区进行测序。16S rDNA包含有9个可变区,分别是v1~v9(ref)。通过提取样品的总基因组DNA,利用细菌16S rDNA V3~V5区(586 bp)的通用引物进行PCR扩增。PCR产物在2%琼脂糖凝胶电泳结果如图1所示,所有的样品能扩增出PCR条带,位于750~500 bp。

1~30:NS患者样本;31~38: 正常人样本图1 PCR电泳结果
2.2样品测序结果及测序深度 Miseq测序得到的PE reads首先根据重叠关系进行拼接,同时对序列质量进行质控和过滤,区分样本后进行OTU聚类分析。统计显示测序读长主要集中在420~460 bp,每个样品中的有效序列数量为30 000多至40 000多不等。为了检测本次测序深度,我们通过随机抽样法分析了各样品的稀释曲线。结果如图2所示,测序的38个样品的稀释曲线都已趋于平缓,表明本次测序的深度基本达到要求,再增加测序的数据量对OTU的发现贡献不大。

图2 测序样品的稀释曲线
2.3肠道菌群物种多样性分析 在所有的样品中,细菌种类最少的不到100种,最多的不超过300种,进一步统计分析患者和健康组物种多样性,结果表明NS患者的细菌种类明显低于健康人群,见图3。
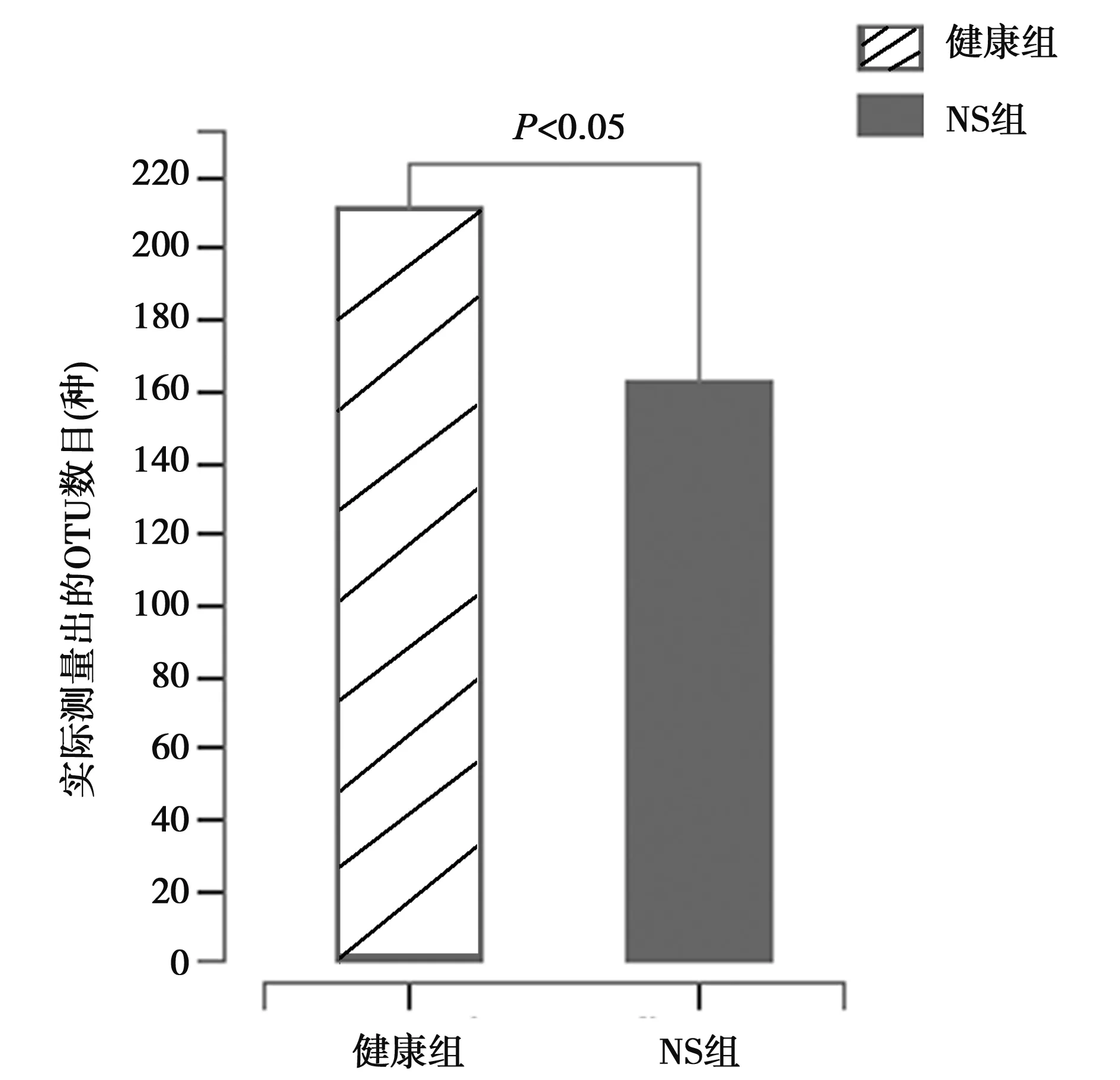
图3 NS组和健康组肠道细菌种类数
2.4NS患者和健康人肠道菌群组成差异分析 基于OTU的物种注释,进一步对NS患者和正常人肠道细菌菌群的群落结构进行统计分析,结果表明NS患者肠道芽孢杆菌纲及乳杆菌目较健康人明显增多;而belta-变形菌纲、假单胞菌科、假单胞菌目、伯克氏菌目、链孢囊菌目、放线孢菌目显著降低。见图4。

A:NS组和健康组不同科的肠道细菌平均所占比例;B:NS组和健康组肠道细菌科级水平图4 NS组和健康组肠道细菌科级水平差异性分析
3 讨 论
正常人肠道菌群的种类有500到上千种,但我们只在每个粪便标本中检测到不超过300种细菌,可能的原因是我们没能检测到很多低丰度的细菌。本实验结果表明:NS组和健康组肠道菌群在门、纲、目、科、属分类水平上都存在统计差异,其中目水平的差异最大;然而在物种水平上,NS组和健康组之间没有统计学差异,主要的原因是组内不同的样品在物种组成上差异很大。
研究表明人体胃肠道微生物的组成在出生后一年内就已建立,一年以后婴儿的肠道菌群就与成人相似并保持稳定,但饮食习惯、用药等都会影响胃肠道微生物的组成〔14〕。其实,正常人肠道菌群差异很大,所以肠道宏基因组的研究需要更大的样本,只有更大的样品、更多的数据才能得到更精确的结论。综上所述,研究结果揭示了NS组和健康组肠道菌群多样性方面有明显差异,菌群结构亦存在较大的差别。该研究结果为将来深入研究某一特定差异菌的对NS发生发展过程中的作用机制打下基础。
猜你喜欢
中老年保健(2022年2期)2022-08-24
军事文摘(2022年16期)2022-08-24
世界科学技术-中医药现代化(2022年2期)2022-05-25
中国饲料(2022年5期)2022-04-26
昆明医科大学学报(2022年3期)2022-04-19
今日农业(2021年11期)2021-08-13
科学导报(2021年29期)2021-06-03
中国生殖健康(2020年4期)2021-01-18
中国生殖健康(2020年4期)2020-12-09
中西医结合肝病杂志(2020年2期)2020-10-27
