
基于分子模拟的低温煤焦油中酚类化合物的溶解特性
2020-01-15 08:29李光升解强张香兰张海永
化工进展 2020年1期
李光升,解强,张香兰,张海永
(中国矿业大学(北京)化学与环境工程学院,北京100083)
基于我国“富煤、贫油、少气”的能源禀赋,煤炭在我国能源利用结构的主体地位短时间内难以改变。低变质程度的煤占到我国全部煤炭储量50%以上,通过热解获取油品和化学品是低阶煤高效清洁化利用的重要方式之一[1-2]。低温煤焦油是低阶煤热解的产物,其中有近1/3 的组分为酚类化合物[3-4]。酚类化合物是重要的化工原料,但存在于焦油中的酚类化合物既影响焦油的稳定性,还增加焦油加氢过程的氢耗、影响加氢产品质量[5],需要将其从低温煤焦油中分离、提取。掌握酚类化合物在焦油中的溶解特性是其高效分离的前提。
目前,有关焦油中酚类化合物的研究大多集中在组成分析[6-8]、分离方法及机理[9-11],对酚类化合物在焦油中的溶解行为及机理研究较少。少量研究对模型焦油体系进行简单热力学数据测定[12-13],也并未将物质结构与溶解特性相关联。此外,对酚类化合物在焦油中的溶解机理鲜见报道。究其原因,一方面因为低温煤焦油的大规模生成和利用是近十几年才出现的事物,另一方面则是由于煤焦油组分的复杂性给实验研究带来的极大困难。近年来,分子模拟方法被广泛应用于宏观热力学性质的计算和微观结构的研究[14],例如,Zhao等[15]和Fu等[16]采用分子模拟的方法,计算比较了不同增塑剂与聚合物间相溶性的强弱,对溶解机理进行了解释;Zeng等[17]借助Extended Flory-Huggins模型与分子动力学模拟(MD),计算多种聚合物之间的相溶性;于共奇[18]基于分子动力学模拟计算石油重质油组分能量、结构等数据,探究了溶解行为及机理。然而,以模拟计算的方法研究酚类化合物在焦油中的溶解特性及机理尚未见报道。
本文采用分子模拟方法从分子水平上对酚类化合物在焦油中结构-溶解特性及机理进行探究,在二元体系中通过酚类化合物与其他组分混合能的计算,将物质结构与酚类化合物在焦油中的溶解规律相关联;在模型焦油混合物中,采用MD方法,借助相互作用能、径向分布函数、电子密度对酚类化合物在焦油中的溶解机理做出解释,以期为酚类化合物的精细化分离提供理论指导和依据。
1 理论基础与模拟计算方法
1.1 理论基础
1.1.1 混合能
在Extended Flory-Huggins 理论中[19-21],吉布斯自由能被描述为式(1)和式(2)。

式中,x为物质的聚合度;φ为物质的体积分数,%;R为气体常数;T为热力学温度,K;χ为相互作用参数;Emix为混合能量,被定义为在相关温度下组分间结合能和配位数的函数,见式(3)。

式中,Z为配位数;Eij为组分i和组分j间的结合能,kJ。
由式(1)~(3)可知,与混合吉布斯自由能类似,混合能是衡量两种物质混溶性质的热力学参数。二元体系混合能的数值越小,表明这两种物质相溶性越强,体系越倾向于混溶状态。
1.1.2 相互作用能
混合物体系的总能量是由化学键能和非键接作用能两部分构成,相互作用能[16]是不同组分非键接作用能量的总和,是衡量混合物中不同组分间相互作用强度大小的重要参数,其计算公式见式(4)。
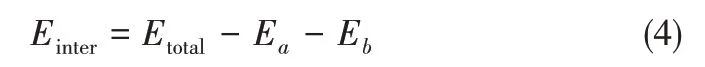
式中,Einter、Etotal分别为组分间相互作用能和体系总能量,kJ;Ea为组分a的单点能,kJ;Eb为组分b的单点能,kJ。通过计算模型焦油中酚类化合物与其他组分间的相互作用能,量化酚类化合物在焦油中受到的缔合作用,进而解释溶解机理。
1.1.3 径向分布函数
径向分布函数[22-23]g(r)是以一个粒子为中心,在距离其r处发现另一粒子的概率密度和该粒子体系随机分布概率密度的比值。通过分析原子间径向分布函数曲线中尖峰的位置,可对分子间相互作用的类型进行判断。研究表明氢键和强范德华力作用范围分别为2.6~3.1Å、3.1~5.1Å(1Å=0.1nm),大于5.1Å为弱范德华力[24]。因此,通过计算苯酚中氧原子(Op)分别与苯的苯环碳原子(Cb)、脂肪烃碳链的碳原子(Ch)、吲哚的氮原子(Ni)、苯并噻吩中硫原子(Sb)的径向分布函数,判断苯酚与其他物质的分子间作用力,进而阐明缔合作用的本质。
1.2 模拟计算方法
1.2.1 混合能计算
运 用Materials Studio (MS) 软 件 内 置 模 块Blends可计算二元体系的溶解行为。从焦油实际组成出发[6-7,25-26],综合考虑物质的结构,以焦油各组分中不同结构的典型化合物为研究对象,模拟计算物质结构对酚类化合物与芳香烃、脂肪烃、杂环氮化物、含硫化合物等焦油典型组分的混合能的影响。各组分中不同结构的典型化合物如表1所示。
模拟细节如下:使用COMPASS 力场[27]来保证混合物热力学性质计算的精确性;物质的模型获取自NIST 数据库(http://webbook.nist.gov/chemistry)以及ChemSpider(http://www.chemspider.com)。对所构建的分子模型进行结构、能量的优化后在Blends模块中输入并计算混合能。
为验证模拟方法的准确性,采用该方法对甲苯、十二烷、苯酚间的混合能进行计算,结果发现混合能从大到小的顺序为苯酚-十二烷(11.90kJ/mol)、甲苯-十二烷(4.80kJ/mol)、苯酚-甲苯(2.36kJ/mol),表明苯酚与甲苯最易互溶,苯酚与十二烷相溶性最弱。模拟计算结果与文献[12]实验规律相符合,因此该方法适用于焦油体系的计算。
1.2.2 焦油模型的构建及MD方法
焦油模型的构建及分子动力学模拟均在MS 软件中完成。在298K、0.101MPa下,借助Amorphous cell模块,采用周期性边界条件,以苯酚、苯、正己烷、吲哚、苯并噻吩这4种焦油中的典型化合物构建焦油模型。焦油模型中各物质分子数及质量比如表2 所示,综合考虑尺寸效应及计算体系大小,焦油模型共包含约3000 个分子,构建好的焦油模型如图1 所示。采用Geometry Optimization 对构建的模型进行结构的初步优化后进行下一步的分子动力学模拟。
MD 模拟细节:在COMPASS 力场下进行分子动力学模拟,选取NPT 系综,模拟温度为298K,压力为0.101MPa,采用Andersen 控温方法和Berendsen 控压方法,对分子间的范德华力和静电作用均采用Group based 方法计算,总模拟时间为100ps,步长1fs,每5000步输出一次轨迹文件,其中前50ps 用于平衡系统,后50ps 用于统计分析相互作用能以及径向分布函数等。

表1 各组分不同结构的典型化合物

图1 焦油模型

表2 模型焦油体系中各物质的含量
在分子动力学模拟过程中,通过衡量体系的温度、能量是否随时间的增长趋于稳定来判断系统达到平衡与否。图2是焦油模型在分子动力学模拟过程中温度和能量的变化曲线,在模拟时间为50ps后,温度变化的标准偏差为0.331%,能量变化的标准偏差为0.261%,表明已经充分达到平衡,因此可对该段轨迹取样并进行热力学性质的分析。

图2 模型焦油体系在分子动力学模拟过程中温度和能量的变化
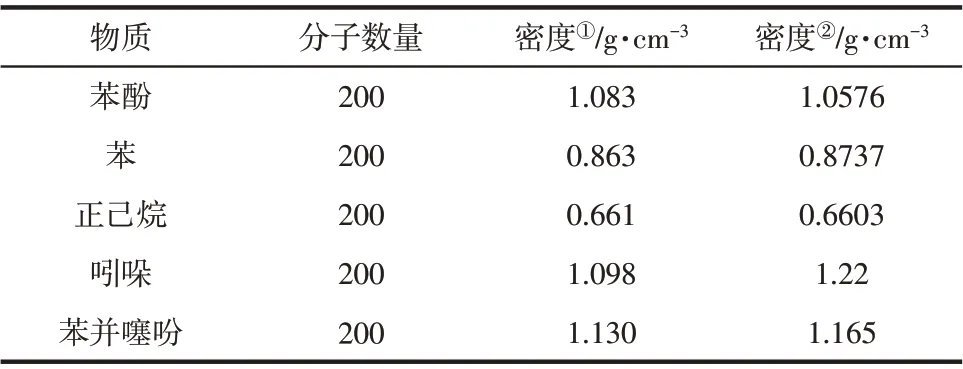
表3 焦油模型中各物质的密度模拟结果与实验值对比
此外,物质密度的模拟计算常被用于验证模型与实际的相符性及模拟方法的可行性[18],因此在该方法下对焦油模型中各物质进行了密度的模拟计算。如表3所示,各物质密度模拟结果与实验得出的数据[28]相差较小,表明该模拟方法较为准确、可行。
1.2.3 氢键分析
在探明氢键是苯酚与吲哚间缔合作用的来源后,借助MS 软件的DMol3 模块,通过平衡体系下苯酚与吲哚的电子密度,分析两者氢键的类型及形成机理。模拟细节如下:采用GGA/VMN-BP 函数在DNP 4.4基组下的密度泛函算法,全局的空间截断为5.5Å,SCF 公差的收敛标准为1.0×10-6Ha,输出的结果文件在DMol3 的Analysis 中分析总电子密度和变形电子密度。
2 结果与讨论
2.1 酚类化合物与焦油各组分的相溶性
2.1.1 酚类化合物结构对相溶性的影响
酚类化合物结构的不同对酚类化合物与各组分物质的混合能的影响如图3所示。苯酚、邻甲基苯酚、邻乙基苯酚、邻丙基苯酚、邻叔丁基苯酚等酚类化合物烷基取代基碳数增加,苯酚、邻甲基苯酚、2,3-二甲基苯酚等酚类化合物烷基取代基数量增加,均导致酚类化合物与苯、正己烷、苯并噻吩之间的混合能减小,表明酚类化合物与这些组分相溶性逐渐增强;而酚类化合物与吲哚的混合能随酚类化合物取代基碳数及数量的增加先减小后增大,在吲哚中的溶解性则先增强后减弱。邻甲基苯酚、间甲基苯酚、对甲基苯酚与其他物质间的混合能变化较小,表明酚类化合物烷基取代基位置对酚类化合物的溶解性影响不显著。此外,当酚类化合物羟基数量增加时,酚类化合物与苯、正己烷、苯并噻吩间相溶性逐渐减弱,而酚类化合物与吲哚之间相溶性未呈现规律性变化。邻、间、对苯二酚与其他组分物质间混合能的差异较小,相比于羟基数量的影响,溶解性的变化并不显著。

图3 酚类化合物结构对混合能的影响
分析原因,酚类化合物烷基取代基碳数及数量、羟基数量的增加均可使其与芳烃、脂肪烃、含硫化合物等组分间的缔合作用增大,使相溶所需能量减小,酚类化合物在这些组分中表现为更倾向溶解;而酚类化合物取代基位置的变化并未使得两者间缔合作用产生显著变化,对溶解性的影响也较小。对于杂环氮化物与酚类化合物的相溶性,酚类化合物烷基取代基碳数的增加也使得这两组分间缔合作用发生变化,相溶性先增强后减弱;而酚类化合物其他结构的变化对两者相溶性未起到规律性的影响。
2.1.2 芳香烃结构对相溶性的影响

图4 芳香烃结构对混合能的影响
芳烃结构的变化对其与酚类化合物混合能的影响如图4所示。苯、甲苯、异丙苯等芳烃,其苯环上烷基取代基的碳数增加,使其与苯酚的混合能呈增大趋势,表明该结构的变化对酚类化合物在芳香烃中的溶解性起到不利影响;对比苯、二甲基苯与苯酚的混合能,烷基取代基数量的增加使得芳烃与酚类化合物相溶性减弱。而当芳香烃苯环数量增加时,苯酚与苯、萘、蒽的混合能先减小后增大,相溶性则先增强后减弱。此外,邻、间、对二甲苯与苯酚的混合能差异较小,三者与苯酚的相溶性差异也不显著。究其原因,芳香烃烷基取代基碳数、数量的增加形成了空间位阻效应,取代基与苯环上电子云发生重叠产生斥力,使芳香烃与酚类化合物的缔合作用减小,混溶所需能量增大,相溶性减弱;苯环数量的增加则使得缔合作用先减小后增大,呈现为相溶性的先减弱后增加;烷基取代基位置的变化对缔合作用影响较小,对相溶性的变化也并不显著。
2.1.3 脂肪烃结构对相溶性的影响

图5 脂肪烃结构对混合能的影响
链状烃碳链长短、直链烃与环烷烃的结构差异对酚类化合物在脂肪烃中溶解性的影响如图5 所示。直链烷烃碳数增加即碳链增长时,混合能逐渐增大,与苯酚的相溶性逐渐减弱;而直链烷烃与环烷烃结构的差异并未使得混合能发现较为明显的变化,相溶性的差异也不显著。分析原因可知,脂肪烃碳链的增长使得其与酚类化合物间缔合作用减小,两者相溶所吸收的能量也增加,表现为对脂肪烃和酚类化合物的相溶产生不利影响。
2.1.4 杂环氮化物结构对相溶性的影响
煤焦油中典型的杂环氮化合物与苯酚的混合能如表4所示。由表可知,杂环氮化物中,吡啶与苯酚混合能值最小,相溶性最强;喹啉、吲哚、咔唑与苯酚混合能逐渐增大,相溶性则逐渐减弱。吡啶和喹啉、吲哚和咔唑,随着苯环数量的增加,形成了空间位阻效应,使缔合作用减小,混合能增大,相溶性也随之减弱。此外,吲哚与咔唑、吡啶与喹啉的含氮五元环、六元环的结构差异,使苯酚在杂环氮化物中的溶解性差异较大,具有含氮六元环的吡啶、喹啉与苯酚相溶性较强。

表4 杂环氮化物结构对混合能的影响
2.1.5 含硫化合物结构对相溶性的影响
表5所示为不同结构的含硫化合物与酚类化合物的混合能。苯酚与苯并噻吩之间混合能最小,与二苯并噻吩的混合能最大。究其原因,苯酚与苯并噻吩结构相似皆具有单个苯环,两者间缔合作用最大,相溶所需能量较少;而苯环数量的增加产生了空间位阻效应,使得二苯并噻吩与苯酚间的缔合作用减小,相溶性变小。因此,随着含氮化合物苯环数量的增加,酚类化合物与含硫化合物的缔合作用先增大后减小,表现为酚类化合物在含硫化合物中的溶解性先增强后减弱。
2.2 相互作用能分析
模型焦油中苯酚与各物质的分子间相互作用能如表6所示,负的相互作用能值表明物质间的相互作用力为吸引力[29]。苯酚受到总分子间相互作用能为57.69×103kJ/mol,主要来源于苯酚与苯、正己烷之间的相互作用,而苯酚与苯并噻吩的相互作用能最小为2.26×103kJ/mol。由前文混合能分析可知,二元体系下,苯酚与其他物质间相互作用力强弱顺序为:吲哚>苯>苯并噻吩>正己烷。然而在多组分的混合体系中,由于不同组分间缔合作用的相互影响,导致焦油模型体系与二元体系不同,苯酚与苯之间的相互作用能最大,其次为正己烷、吲哚、苯并噻吩。

表5 含硫化合物结构对混合能的影响

表6 焦油模型中苯酚与其他物质间相互作用能
2.3 径向分布函数(RDF)及氢键分析
RDF 分析结果如图6 所示。Op与Cb、Sb原子对的径向分布函数曲线分别在3.4Å、3.3Å出现尖峰,表明苯酚与苯、苯并噻吩的缔合作用主要来源于强范德华力,苯酚、苯、苯并噻吩均具有苯环结构,苯酚分别与两者形成了π-π作用。Op和Ch原子对的RDF 曲线在5.1Å 后出现峰值,表明苯酚与正己烷的缔合作用主要由CH-π的堆积作用力贡献。Op和Ni原子对RDF 曲线在3.1Å 出现峰值,属于氢键作用范围,即苯酚和吲哚之间由弱氢键形成缔合作用。究其原因,苯酚、吲哚具有较大偶极矩的O H、N H基团,基团中O、N原子具有电负性导致与该原子成键的氢原子带正电荷,带正电荷的氢原子在空间中与另一个电负性原子接近时则形成了氢键,因此在苯酚与吲哚间形成缔合作用的分子间作用力是氢键。

图6 焦油模型的径向分布函数曲线
此外,苯酚与吲哚间氢键的电子密度分析如图7、图8所示。图7的总电子密度图中苯酚的OH基团与吲哚的NH基团的电子云有明显的接触,该形貌表明苯酚与吲哚两分子之间存在缔合作用,具有电负性的N、O原子形成的氢键为N H···O。图8中的变形电子密度图是基于由苯酚上的O原子和吲哚上的N原子共同存在的截面上形成的,红色表示接受电子的区域,失去电子区域表现为蓝色。图中碳原子图中C 原子周围的红色是接受电子的区域,均处于立体结构下苯环上共价键的中间部分且未发生偏移,属于正常形成共价键的电子云分布。而变形密度切面上体现出O、N原子间由氢键所导致的电子云偏移,围绕着O、N原子接受电子的红色区域发生偏移。吲哚上N H基团中,共价键上电子云分布偏向具有电负性的N原子,氢原子显现出较强的正电性,与苯酚 OH 基团中具有负电性的氧原子具有较强的静电吸引力。因此,在模型焦油体系中,部分可相互接近的苯酚和吲哚分子形成氢键,使两者间产生缔合作用。

图7 苯酚与吲哚的总电子密度

图8 苯酚与吲哚的变形电子密度
3 结论
本文采用分子模拟方法研究焦油中酚类化合物结构-溶解特性,解释溶解机理。本文研究得到的结论如下。
(1)焦油中各组分物质取代基基团、芳环等结构的变化对酚类化合物在焦油中的溶解行为造成显著影响:酚类化合物烷基取代基碳数、烷基取代基数量、羟基取代基数量的增加均促进了酚类化合物在芳香烃、脂肪烃、含硫化合物中的溶解;芳香烃烷基取代基碳数及数量的增加使芳香烃与酚类化合物相溶性减弱;直链烷烃碳链的增长使得其与酚类化合物缔合作用减弱,两者的相溶性减弱;杂环氮化物苯环数量的增加也对其与酚类化合物的相溶起到了不利影响,相比于吲哚、咔唑的含氮六元环结构,吡啶和喹啉的含氮五元环结构使其与酚类化合物具有较大的相溶性;在空间位阻效应的影响下,酚类化合物与含硫化合物的相溶性随苯环数量的增加先增强后减弱。
(2)焦油各组分间缔合作用的互相影响和空间位阻效应是影响酚类化合物溶解特性的关键。酚类化合物与其他组分间缔合作用越大、空间位阻效应越小,相溶性越强。在二元体系中,苯酚与吲哚缔合作用最大,其次为苯、苯并噻吩、正己烷;而在多组分体系中,各组分间缔合作用的互相影响,使各组分与苯酚间缔合作用大小顺序变为苯>正己烷>吲哚>苯并噻吩。
(3)焦油模型中,酚类化合物与各组分物质的分子间非键接作用力形成缔合作用。苯酚与苯、苯并噻吩之间缔合作用的来源是π-π堆积作用力,且苯酚与苯的缔合作用要强于苯酚与苯并噻吩;CH-π堆积作用力、N H···O 氢键是苯酚与正己烷、吲哚的缔合作用力。
猜你喜欢
冶金动力(2022年5期)2022-11-08
武汉工程大学学报(2022年4期)2022-08-26
山东第一医科大学(山东省医学科学院)学报(2021年7期)2021-10-13
西北农林科技大学学报(自然科学版)(2021年1期)2021-03-04
昆明医科大学学报(2020年12期)2021-01-26
人物画报(2020年29期)2020-03-14
人物画报(2020年36期)2020-03-13
化工设计通讯(2020年8期)2020-01-12
鞍钢技术(2018年2期)2018-04-13
分析化学(2017年12期)2017-12-25
