
COL4A5 基因插入突变致Alport 综合征一家系报告
2020-03-12 04:04张莹莹王晶晶毛建华
临床儿科杂志 2020年2期
叶 青 张莹莹 王晶晶 毛建华
1.浙江大学医学院附属儿童医院实验检验中心(浙江杭州 310052)2.浙江大学医学院附属儿童医院肾内科(浙江杭州 310052)
Alport 综合征(Alport syndrome,AS)的发病率为1/10 000~1/5 000,是一种遗传性肾病,其特征为进行性肾衰竭,听力丧失和眼部异常,包括角膜瘢痕、圆锥形晶状体、视网膜变薄和斑点视网膜病[1-3]。AS患者表现出广泛的表型变异性和遗传异质性。实际上,80%~85%的AS 患者的特征是X 连锁的显性遗传,其致病基因是COL4A5;而大约15%的AS患者为COL4A3和COL4A4的致病变异引起的显性遗传或隐性遗传[4]。因此,基因检测在AS临床诊断、预后判断和遗传咨询方面有很大的价值[3]。由于发病率低,流行病学研究报道少见。现报告1个由COL4A5基因插入突变引起的Alport综合征家系。
1 临床资料
先证者,男,11 岁8 个月,5 岁时无明显诱因下出现眼睑、面部浮肿,后渐及双下肢,伴尿量减少,当时无尿频、尿急、尿痛,无腰背部疼痛,无头痛呕吐,无抽搐,无发热咳嗽等。在当地医院诊断为肾病综合征,予以泼尼松治疗(具体不详),尿蛋白++后出院。之后患儿病情反复,在多家医院住院治疗,曾予以吗替麦考酚酯、环磷酰胺、他克莫司等治疗,尿蛋白反复++~+++。于半年前停用激素及免疫抑制剂。两周前患儿感冒后浮肿再发,呈咳嗽、咳痰,伴气喘,无发热、抽搐,无呕吐、腹泻,无头痛,无尿红,无少尿,拟诊肾病综合征收住浙江大学医学院附属儿童医院。入院体格检查:体温36.5℃,心率114次/min,呼吸24次/min,血压134/ 75 mmHg;神清,精神可,柯兴貌,颜面部、双下肢无浮肿,咽无充血,双侧扁桃体无明显肿大;双肺呼吸音粗,未闻及干湿啰音;心律齐,未闻及明显病理性杂音;腹稍膨,肝、脾肋下未及,移动性浊音阴性,双肾区叩痛阴性;神经系统检查无异常。双耳中度神经性听力障碍,双眼视力无异常。实验室检查:尿常规潜血++,尿蛋白++++,红细胞32个/μL。血超敏C反应蛋白<1 mg/L,血常规白细胞计数12.42×109/L,中性粒细胞绝对值7.86×109/L,血红蛋白164 g/L,红细胞压积50.2 %,血小板计数264×109/L;红细胞沉降率25 mm/h;血白蛋白28.3 g/L,白球比例1.29,前白蛋白0.35 g/L,三酰甘油5.64 mmol/L,胆固醇9.41 mmol/L,淀粉酶35.0 U/L;抗核抗体均无异常;TORCH抗体均无异常;T细胞亚群CD3 25.12 %,CD4 4.68 %,CD8 15.02 %,CD4/CD8 0.31;乙肝定性、HIV梅毒、丙肝均为阴性;结核抗体(38 KDa)阴性,结核抗体(16 KDa)阴性,结核抗体(LAM)阴性。24 小时尿量1 300 mL,微量总蛋白3 062.7 mg/L,24小时尿蛋白定量3 981.5 mg;尿蛋白/尿肌酐:1.83。尿培养:无菌生长。胸片未见明显异常征象。腹部B超示胆囊结石。双肾B超示双肾皮质回声增强。骨龄评估示骨龄落后。心电图示正常。双肾磁共振成像(MRI)示双肾外形丰满,右肾下方脂肪间隙异常信号,渗出性改变可能,提示胆囊结石可能,两侧少量胸腔积液。肾穿刺:组织HE染色示肾小管发育不良,泡沫细胞增多,出现幼稚肾小球;荧光染色示肾小球基底膜IV型胶原缺乏;电镜检查示基底膜变薄,厚度约200 nm,系膜增生,系膜区有少量电子致密物沉积(图1)。
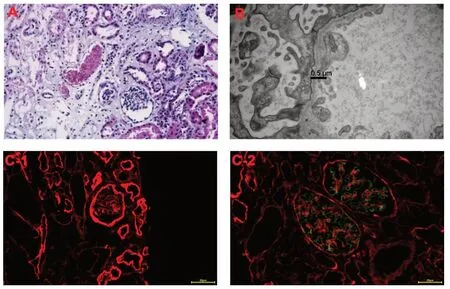
图1 患儿肾组织病理表现
为鉴定该家系的致病突变,经医学伦理审核及家属知情同意进行全外显子组测序和生物信息学分析。结果在先证者的COL 4 A 5基因发现TTCT 插入突变(c.41_42 dup TCTT),并进一步经Sanger测序证实。依照ACMG 临床实践指南该突变鉴定为致病。使用Integrative Genomics Viewer软件分析显示,该插入突变引起移码突变,移码突变导致自插入之后的第13个氨基酸残基开始发生改变,且在第40个氨基酸残基处出现终止密码子,导致蛋白表达提前终止。见图2。先证者的其他家系成员均采用Sanger 测序进行同一可疑致病变异的检测。患儿母亲(II5)和外祖母(I2)均携带该突变基因,分别在35 岁和34 岁时达到终末期肾病(end stage renal disease,ESRD),且两者都有听力受损,但未发现眼部异常。该家系的其他成员中未发现同样的致病变异,表明该变异与家系患病成员存在共分离关联(图3)。

图2 患儿及父母COL4A5 基因测序图

图3 患儿家系图
2 讨论
AS 是由编码Ⅳ型胶原的α 链的基因突变所致。IV型胶原是肾脏基底膜中主要的细胞外基质蛋白,由3条异三聚体三螺旋的α链(α3,α4和α5)组装而成,形成基底膜的三维网络[5-6]。α3链和α4链分别由COL4A3和COL4A4基因编码,都位于2号染色体[7]。而编码α5链的COL4A5基因位于X染色体上[8]。因此,三种典型的孟德尔遗传模式:X染色体连锁显性遗传,常染色体隐性遗传和常染色体显性遗传均可在AS患者中找到[1,9-10]。然而,85%的AS患者为COL4A5基因突变引起的X连锁显性遗传Alport综合征(X-linked dominant Alport syndrome,XLAS)。COL 4 A 5基因位于Xq 22,长约250 kb,含有51 个外显子和51 个内含子,两个已知的剪接变体NM_000495.5 和NM_033380.3分别含有51个外显子的6 513和6 531个碱基,这意味着外显子的整个长度可能对维持蛋白的正常功能都至关重要。COL4A5编码的野生型α5链包含1 685个氨基酸,由氨基末端的26个氨基酸信号肽,1 430 个富含甘氨酸的氨基酸构成的三个关键结构的中间胶原区域(Gly-X-Y)以及羧基末端的229个氨基酸组成[11-12]。
本研究在家系的先证者中检测到COL4A5基因的插入突变(c.41_42in TCTT),且先证者的母亲和外祖母都是该变异的杂合携带者。根据ACMG危险评级和连锁分析证实,这是一个XLAS家系。值得注意的是,家系中受影响的男性患者常常比女性患者具有更为严重的表型。研究发现,通常XLAS 女性患者的肾小球基底膜和Bowman囊中的α5 链发生部分缺失,表现出马赛克表达模式,而男性患者多为Ⅳ型胶原α 5链的完全缺失[13]。其原因可能是女性患者有两条X染色体来平衡基因表达,其中一条染色体随机失活,另一条正常的X 染色体会起代偿作用。相反,由于男性患者只有一条X染色体,因此往往会表现更为严重[14]。此外,之前的研究表明,90%发生截断突变的男性患者将在30岁前进展为ESRD[15-16]。这进一步表明截短突变导致的部分多肽缺失会引起COL4A5基因表达的蛋白功能丧失。
虽然COL4A5基因的截短突变在XLAS中并不罕见,但在中国患者中鲜有报道。先前有报道XLAS 移码突变c.359_363delGTATTinsATAC仅引起中国患者肾脏疾病(未观察到肾外表现)[17],而本研究报道的这个XLAS 家系则有明显的肾外疾病,神经性耳聋。另据报道,在一个中国XLAS家系中发现COL4A5基因的插入突变c.348_349insTCCGG,这一家系除肾脏疾病外,男性患者患有白内障,而非耳聋[18]。这些病例表明COL4A5基因突变引起的XLAS表型多样。
目前,尚无根治AS的方法。临床主要通过减少尿蛋白、延缓肾纤维化等方式来延缓肾衰竭的发生。临床研究表明,血管紧张素转换酶抑制剂和血管紧张素受体阻滞剂对AS 有一定的疗效,且安全性较高[14]。血管紧张素转换酶抑制剂被推荐作为治疗AS的一线用药,如患儿血管紧张素转换酶抑制剂不耐受,则推荐使用二线药物血管紧张素受体阻滞剂[15]。这两种药物均能通过减少尿蛋白,从而使AS 患者发生肾衰竭的时间得以延迟[13]。
综上所述,本研究中的家系先证者患有反复水肿和尿常规检查异常6 年以上,双耳中度神经性听力障碍,基因检测显示COL4A5的TTCT插入突变(c.41_42 dup TCTT)。基因检测有助于XLAS的确诊,本研究扩展了XLAS的变异谱。
猜你喜欢
听力学及言语疾病杂志(2022年5期)2022-09-20
中华实用诊断与治疗杂志(2022年1期)2022-08-31
临床输血与检验(2022年3期)2022-06-22
郑州大学学报(医学版)(2019年3期)2019-06-03
传染病信息(2019年2期)2019-05-17
吉林林业科技(2018年6期)2018-11-21
振动与冲击(2018年4期)2018-03-05
振动与冲击(2017年14期)2017-07-19
听力学及言语疾病杂志(2015年5期)2015-12-24
重庆医学(2015年12期)2015-03-05
