
儿童Alport 综合征1 例COL4A5 基因新发突变分析
2020-03-12 04:04贾实磊高晓洁马颐姣刘俐兵倪芬芬
临床儿科杂志 2020年2期
贾实磊 高晓洁 马颐姣 刘俐兵 倪芬芬 李 俊
深圳市儿童医院肾脏科(广东深圳 518026)
Alport综合征(Alport syndrome)又称遗传性肾炎,是因编码Ⅳ型胶原蛋白α3、α 4、α 5 链基因突变所致的遗传性肾脏疾病,在临床上表现为血尿、蛋白尿及进行性肾衰竭,部分患者合并耳聋和眼部改变[1]。文献报道,Alport综合征患儿X连锁显性遗传占85%,常染色体隐性遗传10%,常染色体显性遗传约5%[1-2]。近年来随着分子诊断技术的快速发展,Alport综合征的诊断从最初依据临床表现,到肾脏组织肾小球基底膜病理,再到基因检测,诊断更精准。本文回顾分析1例不明原因血尿、蛋白尿伴肾功能损害,进展为慢性肾脏病尿毒症期患儿的二代测序及肾脏病理检查结果,探讨Alport综合征患儿的临床特点及基因变异。
1 临床资料
患儿,男性,11岁,体质量25kg。4年前因“发热伴咳嗽5天”入院,查尿常规尿比重1.010,蛋白++,红细胞500/μL。诊断急性支气管炎,急性肾炎综合征,不明原因血尿、蛋白尿,肾功能轻度异常(肌酐85 μmol/L)。建议完善肾活检肾脏病理检查,定期随访。家属拒绝肾活检,亦未再随访。2018年7月,患儿因“呕吐2天,精神萎靡1天”入院。查肾功能肌酐1 051μmol/L,促红细胞生成素1.2 mIU/mL,血红蛋白(Hb)70 g/L。泌尿系超声示双肾体实质回声明显增强,肾皮质髓质分界不清,双侧输尿管无扩张。诊断慢性肾功能衰竭。患儿近1 年余夜尿增多、乏力,无发热、头晕、头痛、恶心呕吐,无视物模糊,面色苍白无明显加重。患儿父母非近亲结婚,无类似家族史。实验室检查:血常规Hb 70 g/L,血小板(PLT)179×109/L;C 反应蛋白(CRP)0.5 mg/L;血尿素氮38.5 mmol/L,血肌酐1 051.5 μmol/L。计算肾小球滤过率(glomerular filtration rate,GFR)5.1 mL/(min·1.73 m2);尿常规蛋白++。诊断慢性肾脏病5期(尿毒症期)。通过多频稳态听觉诱发电位和眼底检查,患儿无高频听力受损,无视网膜脱素改变。肾穿刺活检:电镜下,慢性硬化性肾小球病变,弥漫性肾小管萎缩(萎缩面积超80%);肾脏组织免疫荧光染色α3、α5(Ⅳ)基底膜染色阳性(图1)。患儿经过血液透析肾脏替代治疗半年,2019年2月顺利进行肾移植治疗,口服免疫抑制剂他克莫司,规律随访,目前已上学。
为了进一步明确诊断,经医院伦理委员会审核、家长知情同意后,分别抽取患儿及父母外周血2 mL,送北京金准基因科技有限责任公司检测。提取患儿DNA,并扩增建立含目标基因的全外显子文库,利用液相捕获试剂盒捕获目标基因,然后高通量测序(平均深度不小于200 X),找出相关可能致病基因,用Sanger 测序验证患儿父母相应的突变。结果显示,患儿COL4A5基因存在一处半合子突变c.2631dupA(插入突变),导致氨基酸改变p.G878Rfs*20(移码突变-20位后终止)。由于该突变导致蛋白翻译提前终止,对蛋白功能的影响可能较大。经家系验证,其父母均未携带,为新发突变(图2)。
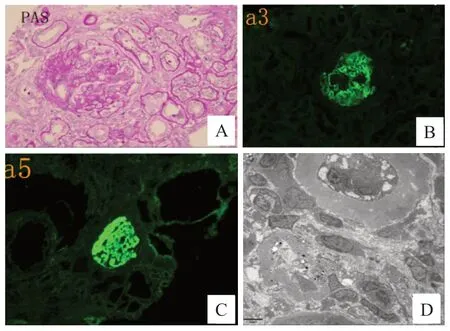
图1 肾组织病理表现

图2 患儿及父母COL4A5 基因测序图
根据美国医学遗传学与基因组学会(ACMG)联合美国分子病理学会(AMP)制订的基因序列变异的解释标准和指南[3]进行致病性分析:①c.2631 dupA突变为移码突变,为无功能变异(非常强致病性证据,PVS1);②c.2631dupA突变为新发突变(De novo突变),父母均未携带该突变,符合X 连锁遗传病特征(强致病性证据,PS 2);③通过检索千人基因组数据库、EXAC数据库,均未见正常人携带c.2631dupA(中等致病性证据,PM2);④患儿临床表现与COL4A5基因突变导致的Alport综合征表型高度吻合(支持性致病性证据,PP 4)。综上,c.2631 dupA 突变的证据强度为“PVS 1+PS 2+PM 2+PP 4“,判断为致病性突变。HGMDpro数据库突变位点c.2631dupA未见报道。
2 讨论
Alport 综合征是儿童最常见的遗传性肾炎,是由于肾小球基底膜的Ⅳ型胶原α 3、α 4、α 5 链的编码基因(COL4A3、COL4A4、COL4A5)突变造成Ⅳ型胶原网形成异常,导致肾小球基底膜结构和功能改变所引起的一类遗传性肾小球疾病,是Ⅳ型胶原肾病的一种。Alport 综合征有3 种遗传类型:X 连锁显性遗传(XLAS)为COL4A5基因缺陷所致,在临床最为常见,约占85%;常染色体隐性遗传(ARAS)为COL4A3或COL4A4基因缺陷所致,约占10%;常染色体显性遗传,不到5%,临床非常罕见[1-2]。
Alport 综合征主要表现为血尿(多为镜下血尿),感音神经性耳聋,眼部异常和进行性肾功能减退。起病隐匿,早期病变表现轻微,仅为镜下血尿,伴或不伴蛋白尿,肾功能异常多在学龄期甚至青春期后出现,听力和视力损害也同样出现较晚。因此在临床上因其他疾病就诊而发现尿检异常进一步确诊Alport综合征的患儿不在少数,临床极易漏诊误诊。本例患儿7 岁时查尿常规提示血尿蛋白尿,血肌酐轻度升高,建议肾活检,但家长拒绝检查。患儿听力、眼底筛查无异常,无肾脏病家族史,加之当时基因检测尚未普及,医师建议其定期随诊尿常规及肾功能,但失访。再次就诊时已是慢性肾脏病5 期,尿毒症期,计算eGFR 5.1 ml/(min.1.73 m2)。肾穿刺活检电镜肾脏病理:慢性硬化性肾小球病变,弥漫性肾小管萎缩(萎缩面积超80%)。肾脏组织免疫荧光染色α3、α5(Ⅳ)基底膜染色阳性。基因检测发现COL4A5基因半合子突变c.2631 dupA(插入突变),导致氨基酸改变p.G 878 Rfs*20,且经家系验证,为新发突变,其父母均未携带,该突变导致蛋白翻译提前终止,对蛋白功能的影响可能较大。依据ACMG指南,其突变变异类型可评级为致病性突变。
约占80%的XLAS 患者有血尿或肾功能衰竭家族史,但本例患儿无阳性家族史。肾组织电镜是确诊Alport综合征的重要手段,典型肾组织病理改变电镜为肾小球基底膜厚薄不均、致密层分层、撕裂、虫蚀样或篮网样改变。但如此典型表现,需要几年或几十年时间,疾病进展到一定程度才能出现。本例患儿肾活检病理亦无Alport 综合征典型电镜表现,因此,即使早期行肾活检检测,仍容易漏诊。文献报道,XLAS肾脏和皮肤Ⅳ型胶原免疫荧光染色大多数表现为α3(-)、α 4(-)和α 5(-)[4],而本例患儿的肾脏和皮肤IV型胶原免疫荧光染色均为阳性。文献报道,有15%~20% Alport综合征患者肾组织α3、α4和α5链分布可以正常,导致Ⅳ型胶原免疫荧光染色阴性,但具体机制不明[5-6]。因此,对于Alport综合征而言,临床表现及肾脏、皮肤的Ⅳ型胶原免疫荧光染色及电镜仅为诊断提供辅助信息,免疫荧光染色阳性仍不能除外Alport 综合征,其确诊最终依靠基因诊断,基因检测是诊断Alport综合征的金标准。
迄今为止,虽已经报道了大约400多种COL4A5基因突变,但目前尚未见有热点突变,错义突变约30%,大多数发生在α 5(Ⅳ)链的胶原区,为甘氨酸残基的替代,被替代甘氨酸的位置或者替代氨基酸本身等均可对蛋白质的三螺旋折叠产生重大影响,导致不同的临床表型[7]。COL4A3、COL4A4基因定位在2号染色体2q35-37上,COL4A5基因定位在X染色体Xq22上。国外研究报道,COL4A5基因致病基因类型不同、位置不同,则临床表型不同,轻重不一。其中突变位于胶原区(外显子1-20)的患者临床表现较轻,肾功能进展最为缓慢,约30岁以后进入慢性肾脏病5期。而携带COL4A5基因突变在第23-51外显子的患者,通常进入肾衰竭的时间较早,或更易合并眼、耳的损害[8-10]。Alport 综合征的病情严重程度还与其他因素有关,如X染色体失活或里昂化(lyonization)、携带修饰基因多态性位点、是否合并其他肾脏疾病及环境因素等[11-13]。本例患儿基因突变发生在COL4A5基因31号外显子,原来的G(甘氨酸)变成R(精氨酸),从突变后的精氨酸起数,第20氨基酸变成终止密码子。移码突变,会导致后续的氨基酸和原来序列的氨基酸不一样,突变对α5(Ⅳ)链蛋白功能影响尚待进一步研究。本例患儿肾衰竭的时间较早,11岁即出现尿毒症,检查未提示有眼、耳的损害。此基因突变位点国内外尚未见报道,补充了人类Alport综合征基因突变数据库,对进一步研究中国人群Alport综合征的发病机制、遗传咨询及产前诊断意义重大。
总之,Alport综合征早期表现缺乏特异性,临床中对于不明原因的血尿蛋白尿,进行性肾功能减退的患儿应密切随访,积极询问家族史。怀疑Alport 综合征的患儿应尽早行基因检测。
猜你喜欢
中国自行车(2018年8期)2018-09-26
中成药(2017年12期)2018-01-19
渔业研究(2018年5期)2018-01-17
中国卫生标准管理(2015年15期)2016-01-15
中国畜牧兽医文摘(2015年9期)2015-12-29
川北医学院学报(2015年5期)2015-12-05
西藏科技(2015年5期)2015-09-26
中国医疗美容(2015年4期)2015-04-27
郑州大学学报(医学版)(2015年2期)2015-02-27
祝您健康(2000年11期)2000-12-31
