
Crystal Morphology of β-HMX Under Eight Solvents System Using Molecular Dynamics Simulation and Experiment
2020-04-20 08:24WANGLeiCHENDongLIHongzhenDUANXiaohuiYUYanwu
含能材料 2020年4期
WANG Lei,CHEN Dong,LI Hong-zhen,DUAN Xiao-hui,YU Yan-wu
(1.School of Environmental and Safety Engineering,North University of China,Taiyuan 030051,China;2.Institute of Chemical Materials,China Academy of Engineering Physics,Mianyang621999,China;3.Southwest University of Science and Technology,Mianyang 621900,China)
Abstract:The crystal morphologies of β-HMX(octahydro-1,3,5,7-tetranitro-1,3,5,7-tetrazocine)in eight pure organic solvents were predicted based on the modified attachment energy(AE)model by using molecular dynamics(MD)method.Results demonstrate that the morphological dominant crystal faces of β-HMX in vacuum are:(0 1 1),(1 1-1),(0 2 0),(1 0 0)and(1 0-2),respectively.The(1 0 0)face is the most polar crystal face and has the largest interaction energy with the solvent molecules,which results in a slow growth rate and appears as dominant face in the final crystal morphology.The(1 0-2)and(0 2 0)faces have the small interaction energies with the solvent molecules,which appear as small areas or even disappear in the final crystal morphology.The order of the aspect ratio of the crystal morphology is:cyclopentanone>cyclohexanone>N,N-dimethylacetamide (DMAC)>pyridine>acetone>triethyl phosphate>propylene carbonate>Dimethyl sulfoxide (DMSO),which indicates that DMSO and propylene carbonate are more favorable for the spheroidization of β-HMX in crystallization experiments.The experimental crystal morphologies of β-HMX in eight pure organic solvents were investigated using a natural cooling recrystallization method.Results show that the predicted morphologies are in good agreement with the experimental results.The attached energy(AE)model is suitable for predicting the crystal morphology of β-HMX,which may serve as a guide in β-HMX recrystallization experiments.
Key words:attached energy(AE)model;β-HMX;crystal morphology;molecular dynamics;simulation
1 Introduction
Crystal morphology is very important in many fields because the different crystal morphologies will determine filterability,drying and flowability,scalability and storability of a material,and can indirectly affect the end-use properties of the material[1],such as formulation.For explosives,additional demands are placed on their hazardous properties,such as impact and friction sensitivity,thermal stability,and charge processing methods.Besides,the crystal morphology also influences the solid content of the charging process[2-5].And these significantly affect the weapon’s final application system performance include functional force,detonation performances,safety,mechanical properties,ballistic performance and etc[6].In general,explosive crystals with a smaller aspect ratio(such as bulk,columnar,and spheroidal crystals)have better diffusivity and low sensitivity,which can significantly improve the safety performance and solid-phase content[6-8].Therefore,the investigation on crystal morphology control of explosives has great value for its further applications.
The crystal morphology of explosives is greatly affected by the crystallization conditions,such as solvents,additives,super-saturation and the growth kinetics during crystallization[9].Among them,the influence of solvent molecules on the crystal morphology is enormous[10], which can be accomplished by experiments and molecular dynamics simulation.Up to now,there are many calculation methods of crystal morphology reported in literatures, such as Bravais-Friedel Donnay-Harker(BFDH)rule[11-13], the periodic bonded chain(PBC)theory[14],Occupancy Model[1]and the modified attachment energy(AE)model[15-20].In calculating the crystal habit of explosive compounds,the modified AE model is the most commonly used and guaranteed prediction method.For example,Han et al[21]..studied the effects of four solvents such as methanol, ethanol, ethyl acetate and acetone on the morphology of CL-20 crystals Song et al[22]..used the attached energy(AE)model to calculate the crystalmorphology of3,4-Dinitro-1H-pyrazole(DNP)in different solvents(ethyl acetate,water,water/EtOH and water/AcOH)All these examples mentioned above illustrated that the AE model is effective in predicting the crystal morphology of explosives[23-24].
HMX is high energy explosive with excellent detonation performance and thermal stability[25],and extensively used as energy ingredient in polymer-bonded explosives(PBXs),solid propellants,and so on.HMX has four solid-phase polymorphs,labeled asα,β,γ,δ-HMX[26].Theβ-HMX is the stable form and most commonly used in industry due to its highest crystal density,lowest mechanical sensitivity.The crystal morphology ofβ-HMX has a great influence on its application and performance.Ilana G et al.[27]obtained prismatic,flake-like and needle-like HMX by slow evaporation in a single or binary solvent.Leif S[28],van der Heijden[29]and Kriber H[3]obtained prismatic HMX inγ-butyrolactone and propylene carbonate by cooling method.In the reported literature on theoretical calculations,Duan et al[30-31].and Yan et al.[32]studied the effect of solvents on the morphology of HMX crystals and successfully predicted the crystalmorphology of HMX in acetone and acetonitrile,respectively.Chen et al.[33]also predicted the crystal morphology of HMX in a mixed solvent such as acetone/γ-butyrolactone,DMF/H2O and acetone-ethyl acetate-water ternary system.There are limited several solvents used for crystallization and lack of the influence relationship of solvents on HMX crystal morphology.However,HMX can be dissolved in many solvents and different solvents have a great influence on the crystals morphology.Therefore,it is very important and necessary to systematically investigate the effects of different solvents on the morphology of HMX,which may guide the modification of HMX crystal morphology and greatly improve efficiency.
In this study,the crystal morphology of HMX in vacuum and eight commonly pure organic solvents with certain solubility,including dimethyl dulfoxide(DMSO),acetone,triethyl phosphate,propylene carbonate,N,N-dimethylacetamide(DMAC),cyclopentanone,cyclohexanone and pyridine,were predicted by the modified attachment energy (AE)model and molecular dynamics(MD)method.The important factors affecting the morphology of HMX crystals are analyzed at the molecular level.Furthermore,the aspect ratios of different crystal morphologies obtained from the simulation results were calculated and compared,and the spheroidization degree of HMX was quantitatively characterized.Finally,the recrystallization experimentofHMX in the above pure organic solvents have conducted.
2 Predicted model and simulation details
2.1 Attachment energy model
The attached energy(AE)model has been widely used to predict the crystal morphology of explosives and medicine[34];it was put forward by Hartman based on the periodic bond chain(PBC)theory[15].The AE model fully considers the crystal unit in the prediction process,and the system energy is more stable.Therefore,the AE model is more accurate than the BFDH method.The attachment energy can be defined as the energy released when the crys-tal flakes is attached to the surface of a growing crystal face,expressed asEatt[19],which can be calculated as Eq(1).The AE model predicts crystal morphology by analyzing the 1/2 symmetry plane distance of the crystal and the distance is related to the linear growth rate of the crystal face,as shown in Eq(2).

WhereElattis the crystal lattice energy (kJ·mol-1),andEsliceis the energy(kJ·mol-1)released by a wafer growth with a thickness ofdhkl.Rhklis the relative linear growth rate of crystal faces(h kl)in a vacuum,which is proportional to its attachment energy.
When the crystal grows in solution,the crystal morphology is affected by the different adsorption interactions between solvent molecules and different faces of the crystal.Therefore,the solvents play an important role in the process of crystal growth and the final crystal morphology.If the stronger the adsorption interaction between the solvent molecule and a certain face of the crystal,the easier the formation of the solvation layer by the solvent molecules adsorbed at the crystal face,therefore,the growth of the corresponding crystal face is suppressed[35].The growth of the crystal faces has to remove solvent molecules adsorbed on the crystal face,and the energy will be lost during the de-solvation process.The attachment energy decreases on the different crystal faces of the crystal,thus,the crystalmorphology cultured in the solution is changed compared with that in the vacuum.The interaction energy between the solvent layer and the crystal faceEintcan be calculated by Eq.(3):

WhereEtotis the total energy of the solvent and the crystal face,Esurfis the energy of crystal face without solvent layer,andEsolvis the energy of the solvent layer without the crystal face.
The binding energy of the crystal face in the unit cell with solvent layer expressed asEs.Escan be introduced as an energy correction term to correct the attachment energy on the vacuum model,which can be calculated by Eq.(4):

WhereAaccis the area of the crystal face(h kl)in the unit cell,andAboxis the total crystal face area of the simulated model along the crystal face(h kl).
We defined the solvent-effected attachment energy aswhich can be calculated according to the following Eq.(5).

The modified morphological theory proposed by Hartman is based on the relative growth rate of each surfaceproportional to the absolute value ofas shown in Eq.(6).

The crystal face with the lowest attachment energy has the greatest influence on the overall morphology of the crystal,because it has the slowest growth rate and becomes the dominant crystal face of the crystal.
2.2 Computational details
All simulations in this work were performed using MaterialsStudio (version 8, AccelrysInc.,USA).The COMPASS force field in the simulation was selected to predict the structure of HMX crystal.The COMPASS force field has proven to be a powerfulabinitio force field that can be parameterized based on extensive experimental data[36].The parameterized COMPASS force field can accurately simulate thermo-physical properties and structure of various condensed-phase materials,especially for energetic materials.
The initial structural file of HMX,used in the calculation of this work,is from the reported literature[37]including the lattice parametersa=6.540 Å,b=11.050 Å,c=8.700 Å and the space groupP21/c.A simple schematic diagram of the entire simulation process taking DMAC and HMX as an example is shown in Fig.1.The molecular and unit cell structures of HMX are displayed in Fig.2a and Fig.2b.Firstly,the initial crystal structure of the HMX was optimized using theForcitemodule in MS.The morphology of the HMX crystal under vacuum condi-tions was simulated by the AE model,and the morphologically important surface(h k l)was obtained.The results show that the crystal of HMX in vacuum has five important faces,which is(0 1 1),(0 2 0),(1 1-1),(1 0 0)and(1 0-2),respectively.The above five important crystal faces were cut out by"cleave"and"super-cell",and the periodic superstructure of each important crystal face of length and width approximately 30 Å was constructed.The operations on the above five important crystal faces are consistent.Secondly,the solvent layers were constructed by the Amorphous Cell module,which had 300 randomly distributed solvent molecules at the target density of each solvent.The size of the solvent box is determined by the size of each periodic superstructure.Third,the HMX crystal face-solvent molecule adsorption model of important face(h kl)was constituted by each important face and the corresponding different solvent boxes.In the established adsorption model,a vacuum layer with thickness 40 Å was created above the solvent layer in order to eliminate the effects of additional free boundaries.The HMX crystal face-solvent molecule adsorption model of each important face(h kl)was optimized using theForcitemodule.Finally,the MD simulations were performed in an NVT ensemble with a temperature of 298℃under the Anderson thermostat for the uniform distribution of solvent molecules.
The calculations for electrostatic non-bond interactions were performed by using the standardEwaldmethod with a calculation accuracy of 0.418 J·mol-1,and the calculation of Van der Waals forces(VdW)is based onatom-basedsummation.The molecular dynamics calculation was carried out on the geometrically optimized HMX crystal face-solvent molecular adsorption model,using an NVT ensemble with a temperature of 298℃.The balanced run was performed with a total simulation time of 200 ps and a time step of 1 fs,during which data was collected every 20,000 steps for subsequent analysis.
3 Experimental section
3.1 Materials and characterization
HMX is a compound with molecular mass of 296.16 and the CAS registry NO.2691-41-0,and its purity is 99.5%,determined by high performance liquid chromatography(HPLC,UltiMate3000DGLC).In this work,The HMX used comes from Institute of Chemical Materials,CAEP.DMSO,acetone and triethyl phosphate used are from Tianjin Chemical Industry Co.,Ltd.Propylene Carbonate,cyclopentanone and cyclohexanone were produced in Chengdu United Institute of Chemical&Reagent.DMAC and pyridine are from Sinopharm Chemical Reagent Co.,Ltd.The purity of all solvents used was greater than 99.0%.The scanning electron microscopy(SEM)images of HMX crystals were obtained using a ZEISS sigma-HD-0129 operated at an acceleration voltage of 3 kV.The power X-ray diffraction(PXRD)patterns were determined by Bruker D8 ADVANCE X-ray powder diffractometer.
3.2 Recrystallization
All of the recrystallized samples of HMX in solvents were obtained by the natural cooling method.Firstly,20 mL of solvent and excess HMX were added to a 50 mL round bottom flask,and the solution was heated from room temperature to 85℃(acetone at 50 ℃)in an oil bath and stirred at 300 r·min-1for 2 h to ensure the formation of a solution mixture containing excess HMX solids.After that,the saturated supernatant was taken out and placed in a 50 mL vial,cooled down to room temperature at a certain rate,subjected to suction filtration and dried at 60℃to obtain the recrystallized samples of HMX.The heated magnetic stirrer(1KIA, RET control-visc)was used to control the temperature and agitation speed of the entire experimental system.
4 Results and discussion
4.1 HMX crystal morphology in vacuum
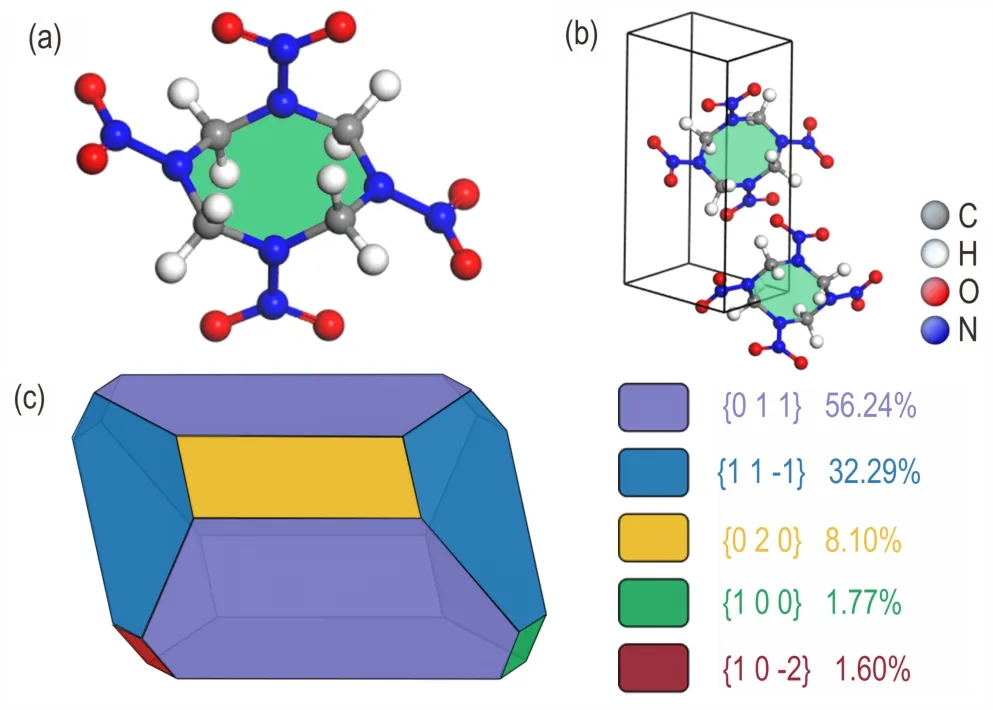
Fig.2 The crystal morphology of HMX in vacuum simulated by the AE model
The crystal morphology of HMX in vacuum was simulated by the AE model,and the parameter information of five important crystal faces(h kl)of HMX ideal crystal was calculated and shown in Table 1.Especially,the area of each important crystal face and the proportion in the crystal are more intuitive as shown in Fig.2.The aspect ratio of HMX simulated by the AE model in vacuum is 1.934,and the aspect ratio reported in the literature is 2.06[30].The difference may be caused by different simulation conditions.In the reported literature,simulations were performed using 200 solvent molecules at a simulation temperature of 276.85 ℃(550 K)and a total simulation time of 140 ps.In this work,300 solvent molecules were used to simulate at a simulation temperature of 298℃and a total simulation time of 200 ps.The results show that there are five important surfaces(h kl)in the ideal crystal of HMX,which are(0 1 1),(1 1-1),(0 2 0),(1 0 0)and(1 0-2).Among them,the morphological importance of the(0 1 1)is the strongest,which has the largest inter-planar distance(d(011)=6.025 Å)and the biggest proportion area in the crystal(Total facet area(011)=56.237%).The absolute value of attachment energy of(0 1 1)is minimum with 110.083 kJ·mol-1,including Van der Waals force(VdW)-83.627 kJ·mol-1and electrostatic potential energy-26.456 kJ·mol-1.And,the(1 0-2)plane is the lowest important crystal face in the crystal,the total facet area is only 1.605% and the minimum growth distance is 4.317 Å.The information of other important crystal faces was listed in Table 1.
Futherly,the surface chemistry of the important crystal faces was analyzed to study the adsorption of solvent molecules on various important crystal faces.And the molecular arrangement and the Connolly surface of the HMX ideal morphology in vacuum was obtained as shown in Fig.3.In Fig.3,the purplegrid on the surfaces indicates the Connolly surface.Obviously,(1 0 0)is the roughest surface because the nitro group are directly exposed and perpendicular to the surface.Secondly,the relatively smooth surfaces are(1 1-1),(0 2 0)and(0 1 1),and their nitro,hydrogen,oxygen and nitrogen atoms are exposed at different positions and angles on the surface.(1 0-2)is the smoothest surface and only the hydrogen atoms are exposed on the surface and the nitro groups are paralleled to the surface.The value ofSis the ratio of the solvent accessible area(Aacc)on the unit crystal face to the corresponding unit crystal face area(Ahkl)[21],which defined as Eq(7).
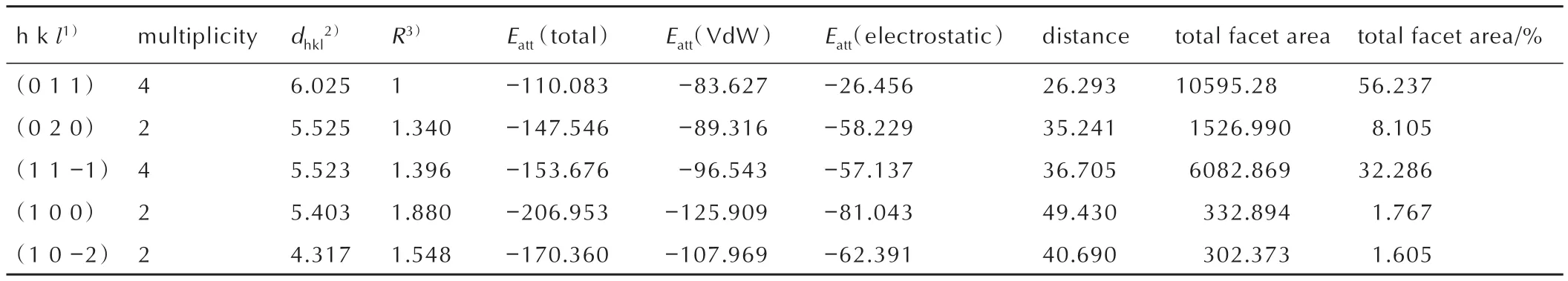
Table 1 Calculate attachment energies for dominant crystal habit faces and other information simulated by AE model in vacuum
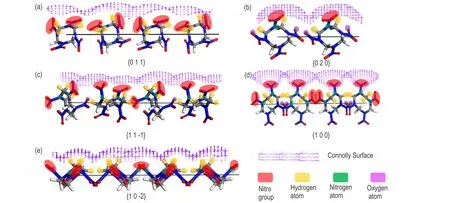
Fig.3 Molecular arrangement and Connolly surface of the crystal face of HMX

The parametersSlisted in Table 2 can quantitatively indicate the difference in the step structures and surface chemistry of the important crystal surfaces.It can be seen from Table 2,theSvalue of(1 0 0)is 1.907,which is the largest among the five important crystal surfaces.This means that the(1 0 0)surface is most exposed to the solvent,and the roughest,which is favorable for the adsorption of solvent molecules.For(1 1-1),(0 2 0),and(0 1 1),theSvalues are not much different.The S value of(1 0-2)is the smallest,which indicates that the surface is the flattest and the proportion of solvent molecules contacted on the surface is the smallest.Based on the above analysis,the relative polarity order of the five important crystal surfaces is:(1 0 0)>(1 1-1)>(0 2 0)>(0 1 1)>(1 0-2).

Table 2 Surface areas for the important crystal surfaces and the value of parameter S1)
4.2 HMX crystal morphology in solvents
The adsorption characteristics of solvent molecules on the important crystal surfaces of HMX can be analyzed from the adsorption behavior of different solvent molecules on a same important crystal surface.Taking(0 1 1)crystal surface as an example,a series of(0 1 1)-solvent molecule adsorption models were constructed with DMSO,acetone,triethyl phosphate,propylene carbonate,DMAC,cyclopentanone,cyclohexanoneand pyridine.The equilibrium configurations of the(0 1 1)-solvent molecule adsorption model structures are simulated as shown in Fig.4.It can be seen from Fig.5,that the solvent molecules are constantly approaching the(0 1 1)surface of the HMX crystal with different distances.This phenomenon is caused by the mutual attraction between the solvent molecules and the polar groups exposed on the(0 1 1)surfacedue to the nitro groups.It can be seen that the binding degree of DMSO molecule to(0 1 1)is the closest,which has the largest interaction energy(Eint=-557.346 kJ·mol-1)and solvent binding energy (Es=-88.889 kJ·mol-1).The negative sign of all mentioned energy indicates the interaction between the solvent and the crystal surface is absorption.The interaction energy(Eint)and the solvent binding energy (Es)of pyridine to the(0 1 1)crystal surface is the smallest,which is 1.297 kJ·mol-1and 0.205 kJ·mol-1,respectively.Table 3 shows that the order of the interaction energy(Eint)and the solvent binding energy (Es)of the(0 1 1)crystal surface with different solvent molecules is:DMSO>DMAC>triethyl phosphate>propylene carbonate>cyclohexanone>cyclopentanone>acetone>pyridine.For the(0 1 1)crystal surface,the solvent binding energy increases with the increase of the polarity of the solvents,and adsorption behavior of the other four surface-solvent molecule adsorption models is basically the same as that of(0 1 1).

Fig.4 The equilibrium configurations of the surface-solvent molecule adsorption model structures after molecular dynamics calculations of(0 1 1)crystal face
The adsorption characteristics of solvent molecules on the important crystal surfaces of HMX can be analyzed from the adsorption behavior of the same solvent molecule on five important crystal surfaces.Taking DMSO as an example,the different adsorption effects of solvents on the five important crystal surfaces of HMX were analyzed.Under the influence of DMSO,the morphology of HMX and the attachment energy of the five important crystal surfaces have changed.The interaction energy (Eint)and solvent binding energy (Es)of(1 0 0)crystal face are the largest,which is-1135.694 kJ·mol-1and-180.467 kJ·mol-1,respectively.This result may be due to the preferential adsorption of polar solvents on polar surfaces.The ideal morphology of HMX in vacuum showed that the(1 0 0)face is the roughest and has the strongest polarity,which results in solvent molecules are preferentially adsorbed at(1 0 0)face.A dense layer of DMSO molecules will be formed on the(1 0 0)plane,which leads to higher energy barriers for HMX molecules in solution to continue to grow on this face.Therefore,the(1 0 0)face will grow slowly and become the dominant crystal face in the final form.The(1 0-2)is the flattest crystal surface,which is not conducive to the adsorption of DMSO molecules,so that solute molecules are easy to attach and grow on this surface.A faster growth rate may cause the surface to eventually disappear.The order of the solvent binding energy(Es)of DMSO molecules to different crystal faces is as follows:(1 0 0)>(1 1-1)>(1 0-2)>(0 1 1)>(0 2 0).The(0 2 0)crystal face has the largest absolute value for the solvent-effected attachment energy (Eatt′),which is 107.964 kJ·mol-1.While the(0 1 1)and(1 1-1)crystal faces have the smallest absolute values,which are 21.193 kJ·mol-1and 21.641 kJ·mol-1,respectively.The order of the absolute values for the solvent-effected attachment energy (Ea′)is as follows:(0 2 0)>(1 0-2)>(1 0 0)>(1 1-1)≈(0 1 1).During the growth process,(0 2 0)and(1 0-2)may be disappeared,which is consistent with the theoretical basis of the AE model,because the relative linear growth rate is proportional to its attachment energy.The relative area size of the important crystal face in the entire crystal morphology is:(0 1 1)>(1 1-1)>(1 0 0).The expected crystal habitcharacteristics ofthe finalmorphology is shown in Fig.5a.The aspect ratio of the predicted HMX crystal morphology in DMSO is 1.838.The comparison result shown in Fig.5a.

Fig.5 Simulation results by AE model and experimental crystal morphology of HMX in solvents
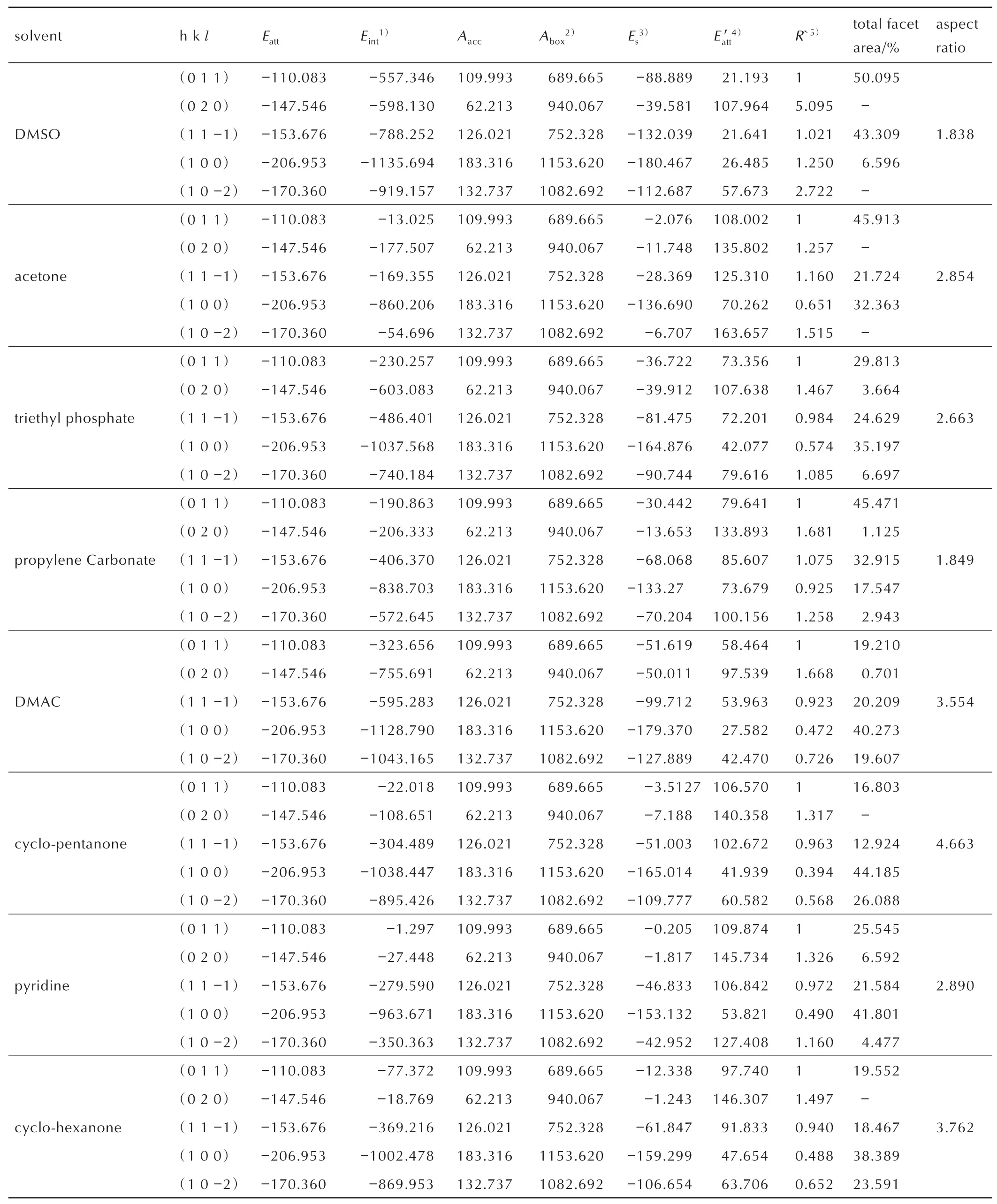
Table 3 Calculated attachment energy for dominant crystal habit faces in different solvents
4.3 Comparison of simulation and experimental results
It can be seen from Fig.4,that the types and strengths of the bonds formed between HMX molecules and solvent molecules are different,and these differences will cause different adsorption effects of the important crystal surfaces and solvent molecules.According to the theoretical basis of the AE model(Section 2.1),the attachment energy of the crystal surface will change with the influence of solvent molecules.The crystal habit characteristics of crystal can be obtained according to the solvent-effected attachment energy.With the increase of the solvent-effected attachment energy,the growth rate of the crystal face increases.Under the continuous influence of the solvent,the crystal face with a rapid growth rate will be submerged,while the slow growing crystal face will be exposed.The final morphology of the crystal will be composed of the crystal face with small attachment energy.The interaction energy(Eint),solvent binding energy(Es),solvent-effected attachment energyand relative growth rate(R′)of other solvent molecules and the five important crystal planes of HMX are listed in Table 3.The negative sign indicates the interaction between the solvent and the crystal surface is absorption.In the Table 3,the interaction energy(Eint)of the(1 0 0)face with various solvent is the highest,while the interaction energy of(1 0-2)with various solvent molecules is relatively weak.Therefore,the dominant crystal faces of HMX are mainly(1 0 0),(0 1 1)and(1 1-1).While(1 0-2)is the smallest proportion area and even disappeared in various solvent.The results of the predicted HMX crystal morphologies in eight solvents are presented in Fig.5.
Recrystallization experiments of HMX were performed in the above solvents and the SEM images of the obtained recrystallized sample are shown in Fig.6,which can be divided into four types:fusiform-like crystal(Fig.5a),rod-like crystal(Fig.5b),needle-like crystal(Fig.5c)and X-like crystal(Fig.5d).
Fig.6 is the power X-ray diffraction of recrystallized HMX samples.The results show that all recrystallized samples areβ-HMX.The prediction results are in good agreement with the experimental samples except for pyridine and cyclohexanone.As shown in Fig.5d,the HMX samples recrystallized from pyridine and cyclohexanone are twinnings.Twinnings are a type of surface defect,while the AE model predicts the morphology of crystal under ideal conditions.Thus the experimental samples recrystallized in pyridine and cyclohexanone are not completely consistent with the predicted results by AE model.The results of theoretical predictions indicate the trend of crystal growth of HMX in solvents and only are used as a reference,because only the effects of solvents and thermodynamics are studied,and these effects factors are very limited.In the actual recrystallization experiments,the kinetic factors and other external factors also play important roles in the morphology of HMX,such as stirring intensity,cooling rate,super-saturation and impurities.
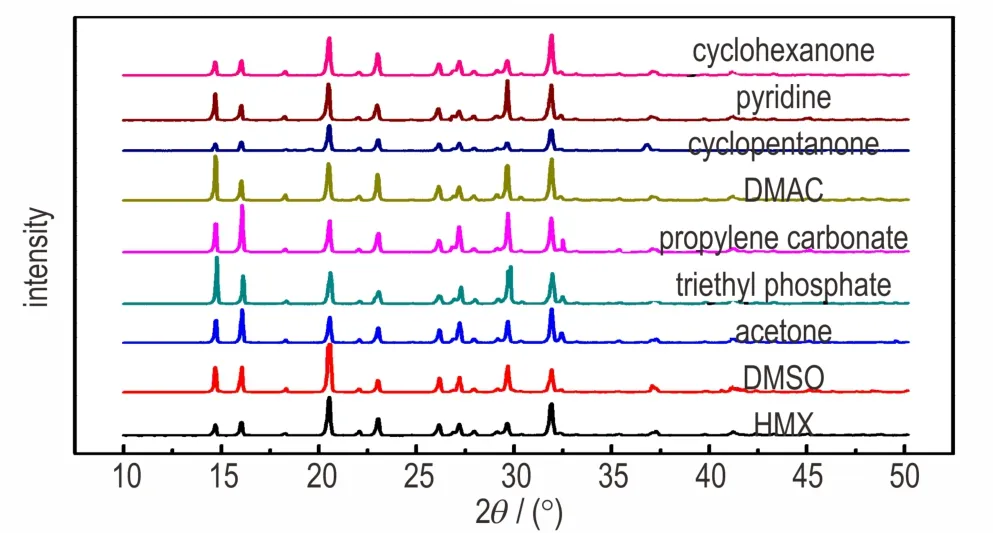
Fig.6 The power X-ray diffraction of the HMX recrystallized in eight solvents
The total facet area(%)of HMX crystal surfaces in vacuum and eight solvents are shown in Fig.7.In general,the crystal morphology of HMX is affected by solvent effects compared with that in vacuum.The area of(0 1 1)face decreased while the area of(1 0 0)face increased in all solvents.The(0 2 0)face area decreases in all solvents and even disappears in DMSO,acetone,cyclohexanone and cyclopentanone.The area of(1 0-2)face increases except for DMSO and acetone.(0 1 1)faces occupy a relatively large area.It can be seen that the crystal morphology of HMX in vacuum and in triethyl phos-phate,propylene carbonate,DMAC and pyridine dominated by five faces,while there are only four faces in cyclopentanone and cyclohexanone,and three faces in DMSO and acetone.For particle morphology,the aspect ratio can be used to determine whether the shape is close to spherical.The aspect ratio is defined as the ratio between the longest and shortest diameters of habit.The aspect ratio value is close to 1,indicating that the morphology of HMX tends to be more spherical.The aspect ratio is the result of the interaction of various solvent molecules with all important crystal faces.The aspect ratio values of the predicted HMX crystal morphology in different solvents are listed in Table 3,and the order of the aspect ratio is:cyclopentanone> cyclohexanone>DMAC>pyridine>acetone>triethyl phosphate>propylene carbonate>DMSO.The smaller the aspect ratio of the crystal morphology,the closer the crystal morphology to the cube,recrystallization of HMX in this solvent is more conducive to the spheroidization.

Fig.7 The total facet area of HMX crystal surfaces in vacuum and eight solvents
5 Conclusion
In this work,the MD simulations of HMX in eight solvents including dimethyl sulfoxide(DMSO),acetone,triethyl phosphate,propylene carbonate,N,N-dimethylacetamide(DMAC),cyclopentanone,cyclohexanone and pyridine systems were performed to obtain the attached energy between the solvent molecules and the dominant crystal face.Different morphologies of HMX crystal were obtained by using the attached energy(AE)model.
(1)The dominant crystal faces of HMX in vacuum are(0 1 1),(1 1-1),(0 2 0),(1 0 0)and(1 0-2).The(1 0 0)face is the most polar crystal face and has the largest interaction energy with the solvent molecules.Hence,it has a slower growth rate and appears as dominant surfaces in the final crystal morphology.And(1 0-2)and(0 2 0)are crystal faces with large attached energy,which appear as the smallest area or even disappears in the final crystal morphology.
(2)The crystal morphology of HMX is affected by solvent effects.The area of different crystal faces is changed with the variation of the solvent.The order of the aspect ratio of the crystal morphology in eight solvents is:cyclopentanone>cyclohexanone >DMAC>pyridine>acetone>triethyl phosphate>propylene carbonate>DMSO.The smaller the aspect ratio of the crystal morphology is in DMSO and propylene carbonate,the closer the crystal morphology is to the cube or spherical and the higher packing density.Recrystallization of HMX in DMSO is more conducive to spheroidization,which will be more beneficial to the subsequent use of HMX.
(3)The recrystallization experimentsresults show thatallofthe recrystallized samplesareβ-HMX.The prediction results are in good agreement with the experimental results.The attached energy(AE)model is suitable for predicting the crystal habit of HMX,which can guide the HMX crystal morphology control by recrystallization experiments.
