
环烷酸酯化反应的热力学研究
2020-04-21 02:43郭银应任玉婷邓昕婷
石油炼制与化工 2020年4期
何 柏,郭银应,连 欣,任玉婷,邓昕婷
(1.重庆科技学院化学化工学院,重庆 401331;2.中国石油西南油气田公司重庆天然气净化总厂)
热力学分析是利用热力学的相关原理和物质的热力学性质,对化学反应和化工过程的发生可能性及其程度等进行计算和预测。虽然影响化学反应的因素很多并且存在各种非理想情况、数据估算误差等,但是热力学分析对于相关反应是否能够发生、反应转化率高低的预判仍然有着非常重要的参考价值[1]。石油酸是一元羧酸混合物,其化合物组分较多,包括脂肪酸、环烷酸以及芳基酸,其中脂肪酸含量较低,环烷酸含量较高,环烷酸质量分数可达90%以上[2-3]。环烷酸是一类带有一个或者多个五元环或六元环的羧酸化合物,其含量在减压馏分油中总的石油酸占比又是相对高的[4],尤其是单环环烷酸、双环环烷酸和三环环烷酸的含量之和可以占到石油酸的85%以上[5-8]。由于石油酸组成的复杂性,其酯化反应因此也非常复杂。黄延召[9]选取了环己基甲酸与乙二醇进行酯化反应并进行了热力学分析,结果表明:在100~197 ℃温度范围内酯化反应焓变大于零,为吸热反应;王艳艳[10]选取了环己基甲酸为模型化合物和乙醇进行酯化反应,热力学分析结果表明,在20~197 ℃温度范围内酯化反应焓变也大于零,为吸热反应;而陈伟东[11]对常一线直馏煤油馏分的酯化脱酸进行研究时,选用了C18直链脂肪酸作为模型酸化合物,研究了脂肪酸与甲醇、乙醇、丙醇、丁醇、乙二醇的酯化反应热力学数据,结果表明,在20~197 ℃温度范围内直链脂肪酸模型化合物与乙二醇的酯化反应焓变小于零,为放热反应。因此,油品中的石油酸种类的不同会使得酯化反应的焓变可正可负,从而会影响反应最佳温度的选择。国外学者[12-13]对脂肪酸酯化反应的热力学研究较多,有关环烷酸酯化反应热力学的研究鲜有报道。为了更好地研究馏分油中的石油酸与醇类酯化反应的热力学情况,分别选取带有五元环和六元环的单、双和三环的6种环烷酸分子[14]为代表模型,研究其分别与酯化反应常用的甲醇、乙醇以及乙二醇进行反应的热力学性质,以更好地探讨环烷酸与醇类反应的热力学规律。
1 环烷酸模型化合物的酯化反应
选用R1~R6共 6种环烷酸为酯化反应的模型酸类化合物(见表1),选用甲醇、乙醇和乙二醇进行酯化反应,其主要反应方程式见式(1)~(3)。
RCOOH+CH3OH=RCOOCH3+H2O
(1)
RCOOH+CH3CH2OH=RCOOCH2CH3+H2O
(2)
RCOOH+HOCH2CH2OH=RCOOCH2CH2OH+H2O
(3)

表1 环烷酸模型分子结构式
由于基团贡献法中的官能团参数是用不完备的试验数据(实际也不可能得到完备的试验数据)根据经验拟合出来的,并且官能团之间的相互作用方式也不太清楚,拟合过程中又会丢失部分信息,所以基团贡献法的计算结果与实际情况往往有较大的差距[15]。鉴于基团贡献法的不足,因此本课题采用Gaussian 09程序包[16]对环烷酸模型分子进行酯化反应相关计算,以探寻相关规律。DFT/B3LYP作为计算方法已经被证明是一种可获得准确电子结构的有效且经济的手段[17-18],因此选用DFT/B3LYP方法[19-20],在6-31G基组水平上对体系中的各原子的几何构型进行了优化和频率计算。
2 热力学分析
考虑到馏分油的黏度随着温度的增加而降低,并且较高的温度有利于分子的热运动,从而动力学上有助于反应的进行,所以绝大部分文献报道的酯化反应温度皆高于373.15 K。但是,反应温度太高又会使得油品发生严重的热解反应造成收率损失,并且产生的烯烃也会影响油品的安定性,所以一般酯化反应的温度低于573.15 K。本研究选择423.15~563.15 K温度范围对环烷酸酯化反应的热力学参数进行计算和探讨。其中,考虑到溶剂分子极化效应,采用PCM模型,同时计算频率,用于确定所找到的驻点是能量极小点(无虚频)[21-22]。采用DFT/B3LYP方法,在6~31 G基组水平上计算了环烷酸酯化反应体系中的6种模型分子以及3种醇在423.15~563.15 K条件下的生成焓、熵和吉布斯自由能,并计算出不同温度下反应的焓变(ΔH)、熵变(ΔS)、吉布斯自由能变(ΔG)。
2.1 各酯化反应的焓变
在423.15~563.15 K温度范围内,环烷酸模型化合物R1~R6与甲醇、乙醇和乙二醇酯化反应的焓变与温度的变化关系如图1所示。由图1可知,酯化反应中的不同反应物及醇类之间的反应焓变存在一定的偏差。在423.15~563.15 K温度区间内,甲醇、乙醇及乙二醇与环烷酸模型化合物进行酯化反应时,R1分别与甲醇、乙醇及乙二醇反应的焓变均大于零,为吸热反应,因此较高的反应温度有利于这些酯化反应向生成酯反应的正方向移动[23];R2分别与甲醇、乙醇及乙二醇反应的焓变为均小于零,表明这些反应为放热反应,根据勒夏特列原理可知,在该温度区间较低温度有利于这些酯化反应向生成酯反应的正方向移动;模型化合物R3、R4分别与甲醇、乙醇及乙二醇反应的焓变均为正值,为吸热反应。双环环烷酸R5与乙醇反应的焓变为负值,为放热反应,因此在该温度区间较低的温度更加有利于反应的进行;R5分别与甲醇、乙二醇反应的焓变为正值,但均小于R1,R3,R4分别与甲醇、乙二醇反应的焓变。三元环环烷酸R6分别与甲醇、乙醇及乙二醇反应时的焓变均大于零,为吸热反应。

图1 反应温度对酯化反应焓变的影响■—R1—甲醇; ■—R1—乙醇; ■—R1—乙二醇; ●—R2—甲醇; ●—R2—乙醇; ●—R2—乙二醇; ▲—R3—甲醇; ▲—R3—乙醇; ▲—R3—乙二醇; 甲醇; 乙醇; 乙二醇; 甲醇; 乙醇; 乙二醇; ◆—R6—甲醇; ◆—R6—乙醇; ◆—R6—乙二醇。图2~图3同
2.2 各酯化反应的熵变
在423.15~563.15 K范围内,R1~R6分别与甲醇、乙醇和乙二醇酯化反应的熵变随反应温度的变化关系如图2所示。由图2可知,随着反应温度的增加,熵变总体逐渐增大。根据热力学第二定律,在孤立体系中发生的任何变化或化学反应,总是向着熵值增大的方向进行,即随着反应温度升高,分子碰撞加剧,反应物与生成物的熵值均增大。在孤立体系中,最终熵变趋近于0,达到反应平衡[24]。R1~R6分别与甲醇、乙醇及乙二醇反应的熵变都趋近于0,这表明在标准大气压条件下,该温度区间内的各反应平衡受温度的影响不大。

图2 反应温度对酯化反应熵变的影响
2.3 各酯化反应的吉布斯自由能变

图3 反应温度对酯化反应吉布斯自由能变的影响
在423.15~563.15 K范围内,R1~R6分别与甲醇、乙醇和乙二醇酯化反应的吉布斯自由能变随温度的变化关系如图3所示。由图3可知,在423.15~653.15 K温度范围内,6种环烷酸模型分子与不同醇发生酯化反应的吉布斯自由能变皆较小。其中,R5与甲醇以及R2与甲醇、乙醇和乙二醇4个酯化反应的吉布斯自由能变均为负,这表明从热力学上来看以上酯化反应能够自发顺利进行;而其余反应的吉布斯自由能变相对更大,酯化反应也会相对困难一些,从而需要通过提高反应温度等手段以使上述反应能够在实际中顺利进行。
3 环烷酸酯化反应的前线轨道分析
为了进一步研究环烷酸模型与醇类发生酯化反应的微观机理,对反应物分子的前线轨道分布进行研究以对酯化反应的机理进行探索,并与前述热力学计算结果进行对比。计算软件、方法以及基组与前述热力学计算一致。
分子的前线轨道也即是LUMO轨道(最低未占据轨道)和HOMO轨道(最高占据轨道)。HOMO轨道是已占据电子的分子轨道中能量最高的,该轨道上的电子所受的束缚力最小,所以在这一轨道中的电子性质极为活泼,更加容易失去电子;而LUMO轨道是所有未占据轨道中轨道能量最低的,从而最容易接受电子。前线轨道在化学作用的时候最容易产生相互作用或者在化学成键反应中起着非常重要的作用。为了验证和比较前面的热力学结果,以R2,R4,R5为例并考虑甲醇、乙醇和乙二醇,分别计算以上化合物的分子前线轨道,结果如表2所示。

表2 部分环烷酸模型分子和醇分子的前线轨道示意
注:各分子中灰色圆球代表碳原子、白色圆球代表氢原子、红色圆球代表氧原子;红色为正电子云,蓝色为负电子云。下同
通过研究环烷酸模型分子R2,R4,R5与甲醇、乙醇以及乙二醇的分子前线轨道能级,分析R2,R4,R5与低分子醇之间的相互作用,结果见图4。由图4可知,甲醇、乙醇和乙二醇都以OH基团通过氧的孤电子对与环烷酸结合。通过前线轨道之间能量差的比较发现,E(LUMO,R2)-E(HOMO,甲醇/乙醇/乙二醇)和E(LUMO,R4/R5)-E(HOMO,甲醇/乙醇/乙二醇)接近,但是R2的HOMO轨道能级高于R4和R5的轨道能级,因此E(HOMO,R2)-E(LUMO,甲醇/乙醇/乙二醇)之间的能量差小于另外两种环烷酸与醇分子之间的能量差。由于反应分子的前线轨道能量越接近,相互之间反应作用越强,所以R2相对更加容易与甲醇、乙醇、乙二醇发生酯化反应,这也与前期的热力学分析结果相吻合。
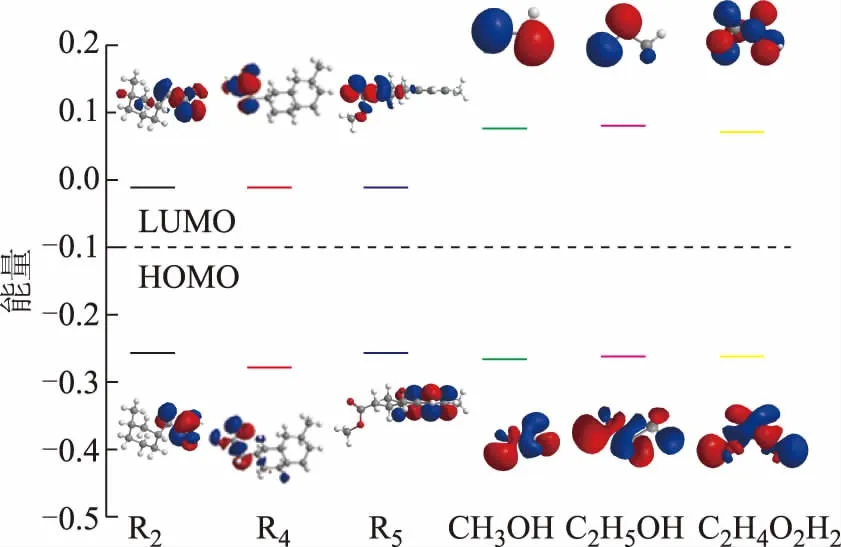
图4 环烷酸与醇的轨道能级分析
4 结 论
(1)采用Gaussian计算软件对6种环烷酸模型化合物与甲醇、乙醇以及乙二醇的酯化反应体系进行了热力学计算,得到了各物质的焓变、熵变以及吉布斯自由能变。
(2)在423.15~563.15 K温度范围内,R2和甲醇、乙醇以及乙二醇反应时的焓变都为负值,R5与乙醇反应的焓变为负值,以上4个反应皆为放热反应,较低的反应温度有利于反应的进行;而其他环烷酸模型分子与甲醇、乙醇和乙二醇的酯化反应皆为吸热反应。环烷酸模型化合物R1~R6分别与甲醇、乙醇和乙二醇反应的熵变都趋近于0,这表明在标准大气压下,该温度区间内的各反应平衡受温度影响不大。不同醇与不同环数环烷酸反应的吉布斯自由能变均不同。其中,单环的环己烷酸R2比环戊烷酸R1更容易与不同醇类发生酯化反应并且自发进行,其吉布斯自由能变从小到大的顺序为:甲醇<乙醇<乙二醇;而其他环烷酸模型分子与不同醇发生酯化反应的吉布斯自由能变更大,所以酯化反应也相对困难一些,从而需要通过提高反应温度等手段以使上述反应能够在实际中顺利进行。因此,较高的反应温度有利于环烷酸酯化反应的进行。
(3)分子前线轨道能级分析表明,醇的OH基团通过氧的孤电子对与环烷酸分子结合,并且R2在一定程度上更加容易与低分子醇发生酯化反应。
猜你喜欢
中学生数理化(高中版.高考理化)(2022年5期)2022-06-01
煤炭与化工(2022年1期)2022-03-19
能源化工(2021年6期)2021-12-30
冰雪运动(2021年2期)2021-08-14
中学生数理化(高中版.高考理化)(2021年5期)2021-07-16
酿酒科技(2021年5期)2021-06-06
上海理工大学学报(2020年5期)2020-11-21
电子制作(2016年19期)2016-08-24
云南中医学院学报(2015年1期)2015-07-31
创新科技(2014年16期)2014-12-22
