
接触图辅助的过程重采样蛋白质构象空间优化算法
2020-05-12 09:09李章维余宝昆周晓根张贵军
小型微型计算机系统 2020年3期
李章维,余宝昆,胡 俊,周晓根,2,张贵军
1(浙江工业大学 信息工程学院,杭州 310023)
2(密西根大学 计算医药与生物信息学院,美国密西根州安娜堡 48109)
E-mail:zgj@zjut.edu.cn
1 引 言
作为人体内分布最广、功能最复杂的一类生物大分子,蛋白质在人体的生命活动中发挥着极其重要的作用.研究蛋白质的结构是研究其生物功能及活性机理的基础,并且对新蛋白的发明和药物标靶蛋白的设计具有十分重要的指导意义[1].传统实验测定蛋白质结构的方法(如:X射线晶体衍射法、核磁共振成像与冷冻电镜等)所需时间长且花费巨大,无法满足大规模蛋白质结构测定的需求[2,3].因此,从蛋白质的一维氨基酸序列出发,直接预测蛋白质三维结构是结构生物学领域的一个重要问题[4].
自由模板(FM)是蛋白质结构预测领域的最具挑战性的一类研究课题.对于任意一条蛋白质的氨基酸序列,其可能形成的空间结构数目是极其巨大的,现有的计算资源难以在巨大的构象空间中搜索到能量最低的构象[5].针对蛋白质构象空间优化问题,研究者们提出了很多构象空间优化算法,如进化算法[6-10]、蒙特卡洛算法[11-13]、副本交换算法[14-16]、构象空间退火[17]等方法.为了进一步减小构象搜索空间,基于片段组装策略[18]的构象空间搜索在从头预测中得到广泛应用,其主要代表有Rosetta[19-21],Quark[22],STRATCH[23],PROFESY[24],FRAGFOLD[25]等一系列方法,特别是Rosetta和Quark方法在国际蛋白质结构预测竞赛(CASP)[26,27]中表现突出.
为了提高蛋白质结构预测精度,CONFOLD[28,29]使用预测的二级结构和残基接触[30]转化成空间约束,然后使用这些空间约束构建蛋白三维结构模型.Filb-Coevo[31]使用残基接触图约束产生高质量的片段库[32,33],进而使用片段组装方法搜索构象.RMA[34]算法在遗传算法的框架下使用预测的二级结构增强对构象采样空间的探索.DPDE[35]算法使用距离谱[36]指导差分进化进行蛋白质结构预测.SCDE[37]算法使用基于二级结构和残基接触的选择策略指导构象空间采样.在进化计算框架下[38-40],RMA[34]、DPDE[35]、SCDE[37]算法使用蛋白质结构的先验知识辅助蛋白质结构预测,有效地提升了蛋白质结构预测精度.除了蛋白质结构的先验知识,种群更新中的过程信息同样十分重要,由于能量函数的不精确,在种群进化的过程中,一些结构合理但能量较高的构象可能会被淘汰.
为了保留结构合理构象的片段信息达到指导种群进化目的,提高蛋白质结构预测精度,本文提出了一种接触图辅助的过程重采样蛋白质构象空间优化算法(CMPR).在种群更新的每一代,首先,根据残基接触打分模型选择结构合理的构象,然后根据选出的构象构建过程片段库,基于过程片段库使用过程重采样策略指导种群变异.12个测试蛋白的实验结果表明,CMPR方法缩小了构象搜索空间,增强了对近天然态构象区域的采样,相对于Rosetta和Quark方法提升了蛋白质结构预测的精度.
2 算法设计
2.1 过程片段库构建
在采样蛋白质构象的过程中,从采样过程构象中提取出来的片段库称之为过程片段库.图1所示是目标序列第一个位置的3~9残基过程片段库的构建.对于序列长度为L的N个蛋白质构象,记录N个构象第一个3~9残基窗口内的二面角(φ,φ)构成3~9残基片段库,然后窗口向下滑动记录第二个位置的3~9残基片段库,直到构建目标序列所有位置上的过程片段库.
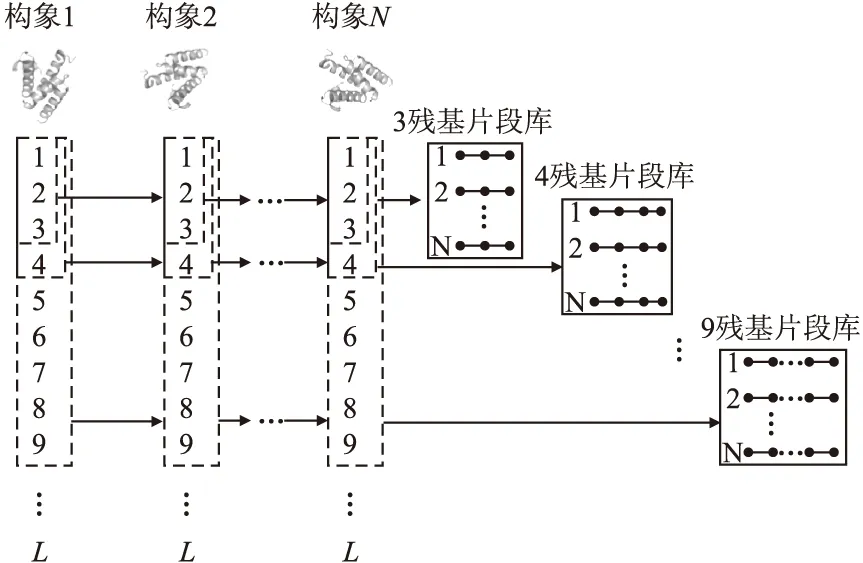
图1 构建过程片段库
2.2 残基接触打分模型
当蛋白质序列上两个残基间Cβ-Cβ(甘氨酸Cα-Cα)的空间距离小于8Å,就认为这两个残基之间有残基接触[41].为了在种群更新的过程中提取构象的片段信息,本文设计了残基接触的打分模型来选择构象构建过程片段库.根据目标蛋白序列,使用RaptorX-Contact[42,43]预测目标蛋白的残基接触图,使用如下公式计算蛋白质构象的残基接触得分:
(1)
(2)
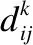
根据设计的残基接触打分模型,如果一个蛋白质构象的残基接触得分较低,换言之,这个蛋白质构象的残基接触和预测的残基接触很符合,那么就称这个蛋白质构象的整体结构是相对合理的.
2.3 过程重采样策略
为了使用过程片段库指导种群变异,本文设计了过程片段库的片段组装策略.对一个构象进行片段组装之前,首先使用Rosetta的能量函数score3[19]计算构象的能量E,公式(1)和公式(2)计算构象的残基接触分数Scon,然后在构象上随机选择一个长度为[3,9]残基长度的片段插入窗口,从窗口对应位置和片段长度的过程片段库中随机选择一个片段,使用该片段中的二面角φ和φ替换当前构象中的二面角.当片段替换完成后,重新计算片段插入后构象的能量E′和残基接触分数S′con,根据如下条件判断是否接收片段插入:
a)若E′-E≤0,S′con-Scon≤0,则接收这次片段插入;
b)若E′-E≤0,S′con-Scon>0,则根据概率Pcon判断是否接收,Pcon公式如下:
(3)
kT为温度常数;
c)若E′-E>0,S′con-Scon≤0,则根据概率Pe判断是否接收,Pe公式如下:
(4)
d)若E′-E>0,S′con-Scon>0,则拒绝这次片段插入.
若连续拒绝插入次数达到最大连续拒绝次数Rmax,令kT=kT+s,s表示升温步长.
2.4 交叉操作
图2所示为个体xi交叉操作示意图,对于个体xi,首先从种群中随机选择一个个体xj,xi≠xj,然后在xj上随机选择一个3残基长度的片段,使用这个3片段上的二面角替换xi相同位置片段上的二面角,得到交叉后的个体x′i,交叉操作增加了种群的多样性.
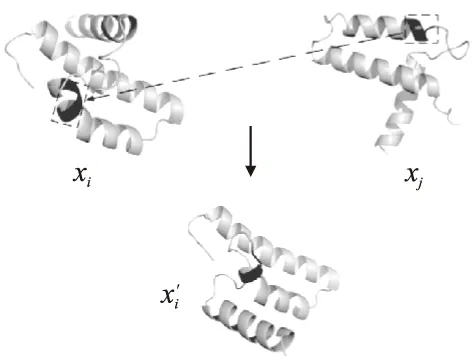
图2 交叉操作
2.5 算法描述:
算法1.CMPR算法流程
输入:目标蛋白序列,片段库,残基接触图
输出:蛋白质三维结构

其中NP表示种群规模,CR表示交叉选择概率,p表示变异选择概率,Q表示片段库提取比例.第1行表示种群初始化过程,对NP个全伸展的构象使用Rosetta的一阶段和二阶段进行初始化.第24行种群选择操作中,把交叉变异前的父代种群和交叉变异后的子代种群合并,使用残基接触打分模型对合并种群个体打分,保留残基接触得分最低的前一半个体进入下一代种群.第25行根据残基接触打分模型从NP个个体中选择残基接触分数最低的Q*NP个体构建过程片段库.
3 实验结果与分析
3.1 测试蛋白和参数设置
实验选择CASP12竞赛中的12个测试蛋白,如表1所示,这些测试蛋白序列长度从39到146,折叠类型包括α、β和α/β类型,所有测试蛋白从Robetta[44]服务器下载去除同源的片段库.本算法在Rosetta平台上实现,使用Rosetta3.4版本的ClassicAbinitio协议.
设置种群规模NP=100,种群更新次数Gmax=100,交叉概率CR=0.5,变异概率p=0.5.最大连续拒绝次数Rmax=150,温度参数初始值kT=2,升温步长s=2,片段库提取比例Q=0.1.通常研究者会在L,L/2,L/5,L/10之间选择使用的残基接触数量[45],L为目标蛋白的序列长度,残基接触数据根据置信度排序,置信度越高排序越靠前,本实验选择前L个残基接触数据计算蛋白质构象的残基接触分数.

表1 测试蛋白集
在CMPR算法运行的过程中,记录除种群初始化阶段之外构象搜索过程中接收的所有构象,使用聚类工具SPICKER[46]对这些构象聚类,记录聚类的第一个类心点作为预测结果.Rosetta方法使用Rosetta默认的参数,保持和CMPR相同的计算代价,记录第三第四阶段蒙特卡洛轨迹接收的所有构象经过聚类得到的第一个聚类中心作为预测结果.Quark的预测结果通过Quark服务器在线提交任务得到5个预测蛋白结构模型,选择第一个模型作为预测结果和其他两种方法比较.
实验采用均方根偏差(RMSD)和TM-score[47]两个评价指标比较目标蛋白的预测结构和天然态蛋白的结构相似度,RMSD值越小表明预测结构与天然态蛋白质结构越相似,TM-score的值在[0,1]之间,TM-score值越大表明预测结构与天然态蛋白质结构越相似.
3.2 结果分析
表2中是CMPR算法与Rosetta和Quark的预测结果的比较.在12个测试蛋白中,CMPR预测结果的TM-score大于0.5的有8个,其中有11个测试蛋白的预测精度在TM-score和RMSD上优于Rosetta,有7个测试蛋白在TM-score和RMSD上都优于Quark.其中测试蛋白1GYZ、1AIL、1GB1、1SAP、1TIG、1BQ9、1WAP在TM-score和RMSD上,CMPR的预测精度比其他两种方法高.CMPR算法的平均TM-score比Rosetta高0.16,比Quark高0.04,平均RMSD比Rosetta低3.13Å,比Quark低1.51Å.总体而言,CMPR算法比Rosetta和Quark预测精度更高.

表2 算法对比结果
为了验证残基接触信息的使用和过程重采样策略的有效性,本文设计了仅使用残基接触信息和仅使用过程重采样策略的对比实验.CM算法把CMPR算法中的变异操作更改为仅使用原片段库进行片段组装.PR算法把CMPR算法中的种群筛选中残基接触打分模型替换成Rosetta的score3,使用score3选择低能量构象构建过程片段库,过程片段库的片段组装中,若E′-E>0,使用公式(4)判断是否接收片段插入,否则,接收片段插入.对比实验结果如表3所示.

表3 CMPR组件比较
对比表2和表3的结果可以看出,仅使用残基接触的CM算法,所有12个测试蛋白的TM-sore比Rosetta高,平均TM-score值比Rosetta高0.11,平均RMSD比Rosetta低1.68Å.仅使用过程重采样的PR算法,12个测试蛋白中9个测试蛋白的TM-score比Rosetta高,其平均TM-score值比Rosetta高0.06,其平均RMSD比Rosetta低0.91Å.CMPR算法平均TM-score比CM算法高0.05,比PR算法高0.1,平均RMSD比CM算法低1.45Å,比PR算法低2.22Å.实验结果表明,仅使用残基接触信息和过程重采样方法相比较于Rosetta都提高了蛋白质结构预测精度,而CMPR结合残基接触和过程重采样策略得到了更高的预测精度.
3.3 算法采样能力分析
部分蛋白的构象采样结果如图3所示,图中横坐标表示构象与天然态蛋白质比较的RMSD值,纵坐标表示构象的能量值.从图中测试蛋白4UEX、1SAP可以看出,虽然CMPR算法总体的构象采样范围比Rosetta小,但是比Rosetta搜索到更多能量更低和RMSD更低的构象区域,因此预测精度更高.1BQ9和1WAP是两个结构较复杂的β折叠型蛋白,Rosetta虽然搜索到了更低能量的构象区域,但是由于能量函数的不精确,低能构象的结构与天然态蛋白质结构相差较大,CMPR算法虽然无法采样到比Rosetta更低能量的构象区域,但是得到的最低能量区域的构象RMSD更小,所以得到结果的预测精度更高.
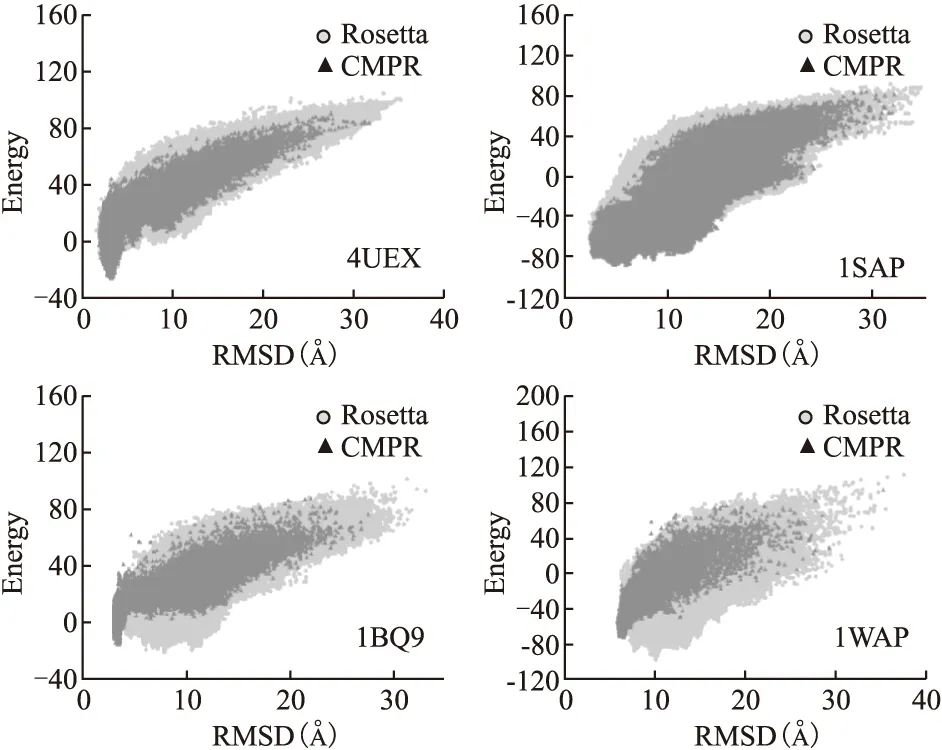
图3 构象空间采样比较
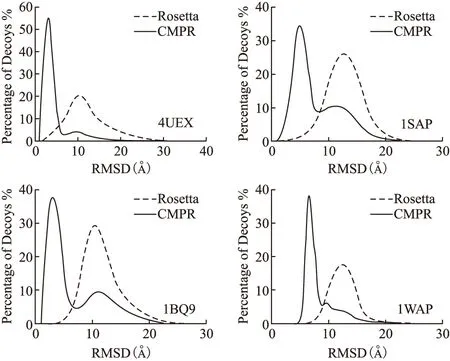
图4 构象RMSD分布
图4所示横坐标表示构象与天然态蛋白质结构之间的RMSD,纵坐标表示每个区域内构象数目所占构象总数的百分比.从图中4个测试蛋白的构象RMSD分布图可以看出,CMPR采样到的低RMSD构象占总构象数目的比例比Rosetta采样的更大,因此CMPR算法比Rosetta有更好的近天然态采样能力.
4 结 论
本文提出了一种接触图辅助的过程重采样蛋白质构象空间优化算法,在进化计算的框架下,基于残基接触图设计了残基接触的打分模型,使用残基接触打分模型筛选种群,提取结构合理构象的片段信息构建过程片段库,结合残基接触图的先验知识和种群进化的过程信息指导构象空间采样.12个测试蛋白的实验结果表明,本文提出的CMPR算法有效地缩小了构象搜索空间,增强了对近天然态构象区域的搜索,有较高的预测精度,是一种有效的蛋白质结构从头预测方法.下一步的研究中,将结合更多的先验知识构建更精准的过程片段库进行蛋白质结构从头预测.
猜你喜欢
今日农业(2022年15期)2022-09-20
生物化学与生物物理进展(2022年7期)2022-07-25
生物化学与生物物理进展(2022年6期)2022-07-21
肝博士(2022年3期)2022-06-30
西南农业学报(2021年10期)2021-12-14
海外星云(2021年9期)2021-10-14
中学生数理化(高中版.高考理化)(2021年2期)2021-03-19
生物学教学(2018年3期)2018-08-08
中学生物学(2018年8期)2018-03-01
中学生物学(2008年6期)2008-08-29
