
乙二胺四乙酸和乙二胺四亚甲基膦酸清除铁垢性能的量子化学计算
2020-07-01 10:43赵晓洋南俊民
华南师范大学学报(自然科学版) 2020年3期
赵晓洋, 唐 静, 刘 伟, 肖 信, 南俊民*
(1. 河南工业职业技术学院环境工程系, 南阳 473009; 2. 华南师范大学化学学院, 广州510006)
在化学清洗工业中,通常采用的无机酸具有挥发性强、气味浓、对设备腐蚀性强等缺点,而有机酸通过螯合作用溶解污垢,具有酸性小、对金属腐蚀性小、无毒无污染等优点. 乙二胺四乙酸 (EDTA)是一种氨基多羧酸螯合剂,能够与Fe、Co、Ni、Cu等金属离子形成稳定且溶于水的配合物,已被广泛应用于工业、农业、医药、科研等领域[1]. EDTA可作为螯合清洗剂,具有安全、高效、工期短等优势[2]. EDTA与污垢中金属离子有很强的螯合作用,可以有效清除钙垢、铁垢、铜垢等,对金属基底腐蚀小,可在金属表面生成保护膜,除垢后无需后续钝化保护过程. 乙二胺四亚甲基膦酸(EDTMP)是一种氨基多膦酸螯合剂,已被广泛应用于水处理. 在水溶液中,EDTMP与Ca2+、Mg2+、Fe3+等可形成松散的螯合物,破坏水垢的正常结晶,有效抑制碳酸盐、硫酸盐、磷酸盐及水合氧化铁的沉淀,可阻止硬垢的生成.
垢样取自平顶山某盐化有限责任公司的列管式钛换热器. 换热室的壳程材料为16MnR钢,管程材料为钛管. 在生产中,卤水物料进入管程被蒸发浓缩,过热蒸汽进入壳程被加热,运行温度为143 ℃. 由于NaCl盐分对蒸汽管道有较强的腐蚀作用,壳程加热室容易结生灰色铁垢,具有铁磁性,主要成分为Fe3O4. 在高温下生成的铁垢非常致密难溶于水,不溶于无机酸溶液. 由于管程使用钛管,不能使用氢氟酸;而无机酸如盐酸、硝酸、磷酸、硝酸+盐酸、硝酸+硫酸均不能溶解铁垢,只能采用有机酸才能溶解铁垢. 而在筛选可用于化学清洗的有机酸时发现:含有膦酸基的羟基乙叉二膦酸(HEDP)效果好[3]. EDTMP含有膦酸基且与EDTA结构相似,通过溶解铁垢实验发现,EDTA可以溶解铁垢,而EDTMP不能溶解铁垢. 虽然已有量子化学计算研究[4-7]报道了EDTA和EDTMP与过渡金属元素生成配合物的相关研究,但是它们与Fe3+配合的分子结构对比研究尚未报道. 使用量子化学计算方法,从分子结构的角度,解释EDTA清洗铁垢的效果强于EDTMP的原因.
1 计算方法
采用Gaussview 6.0软件建立Fe(EDTA)-和Fe(EDTMP)-配合物的分子结构,使用Gaussian 16软件基于密度泛函理论进行计算[8]. 利用B3LYP方法,对所有原子采用DEF2-TZVP基组. 以水为溶剂,在基于密度的溶剂模型(Solvation Model Based on Density,SMD)中,对2种配合物的几何结构进行构型优化和频率计算,计算收敛阈值取程序默认值. 所得结构振动分析没有虚频,对应于势能面上的稳定结构. 对所得结构采用Multiwfn 3.6程序[9]进行波函数分析,包括:几何构型分析、扩展电荷分解分析(Extended Charge Decomposition Analysis,ECDA)、约化密度梯度(Reduced Density Gradient,RDG)分析和拓扑(Atoms in Molecular,AIM)分析. 使用NBO 3.1程序进行自然键轨道(Natural Bond Orbital,NBO)分析. 使用(Visual Molecular Dynamics,VMD)软件[10]对几何构型、RDG、AIM、NBO进行图形化绘制.
2 结果与讨论
2.1 配合物的结合能和扩展电荷分解分析
EDTA具有较强的鳌合能力,其结构中2个sp3杂化的N原子和4个羧基O原子都参与配位,可与大多数金属离子形成物质的量之比为1∶1的配合物. EDTMP与EDTA结构相似,仅有羧基和膦酸基的区别. EDTMP与EDTA都可与Fe3+形成配合物,但EDTA对Fe3+的鳌合能力比EDTMP的强. 采用量子化学方法,有望从2种配合物的分子结构差异来揭示这一现象的本质原因.
对2种配合物在低自旋和高自旋状态下的结构分别进行了优化,配合物的能量和结合能见表1. 从配合物的结合能可以看出2种配合物结合的紧密程度,结合能越大,配体与中心离子结合得越紧密.

表1 配合物的结合能和转移电荷Table 1 The binding energies and charge transfer of the complexes
注:Ec为配合物的能量;El为配体的能量;EFe3+为Fe3+的能量;Eb为配合物的结合能.
分别计算配合物、配体和Fe3+的能量,由公式
Eb=Ec-El-EFe3+
可得配合物的结合能. 可以看出,同种配合物的高自旋态能量比低自旋态能量略低,但同种配合物的低自旋态结合能比高自旋态的大,且结合能的能量差远大于高、低自旋态的能量差. Fe(EDTA)-的高自旋态能量比低自旋态能量低0.816 eV,但低自旋态的结合能比高自旋态的低5.388 eV,Fe(EDTMP)-的高自旋态能量比低自旋态能量低1.061 eV,但低自旋态的结合能比高自旋态的低5.143 eV. 结合能数据说明低自旋态有利于配体与Fe3+的结合. 在高温状态下,Fe3+由高自旋转态变为低自旋态,发生了自旋交叉的现象,并与配体形成配合物[11].
扩展电荷分解分析可以定量描述配体向金属离子的电荷转移数量[12]. 从表1中的转移电荷数据可以看出,同类配合物的低自旋态配体向Fe3+转移的电子数均大于高自旋态的,表明低自旋态更有利于配体向Fe3+转移电子. 在低自旋态下,从EDTA向Fe3+转移的电荷数为2.708 e,从EDTMP向Fe3+转移的电荷数为2.632 e,说明EDTA的转移电荷数比EDTMP的多.
2.2 配合物的分子结构
优化后的低自旋配合物分子结构如图1所示. Fe(EDTA)-的结构由1个EDTA分子的4个羧基和2个N原子参与配位,每个羧基提供1个O原子和2个N原子,与中心Fe3+形成1个六配位的八面体结构. 对于Fe(EDTMP)-,其结构与Fe(EDTA)-的相似,不同的是羧基变成了膦酸基. 尽管EDTA和EDTMP分子与Fe3+形成的配合物结构相似,但二者溶解铁垢的性能存在差异,说明二者分子的差异性起了重要作用. 从表2可以看出,Fe(EDTA)-比Fe(EDTMP)-的在八面体平面位置的对应键长略短. Fuzzy键级反映了2个原子间共享的电子对数. 从键级上可以看出,Fe(EDTA)-的键级比Fe(EDTMP)-同类型键的大. 通过键长和键级的比较可以说明:EDTA与Fe3+结合得更加紧密,这与结合能的结果一致.
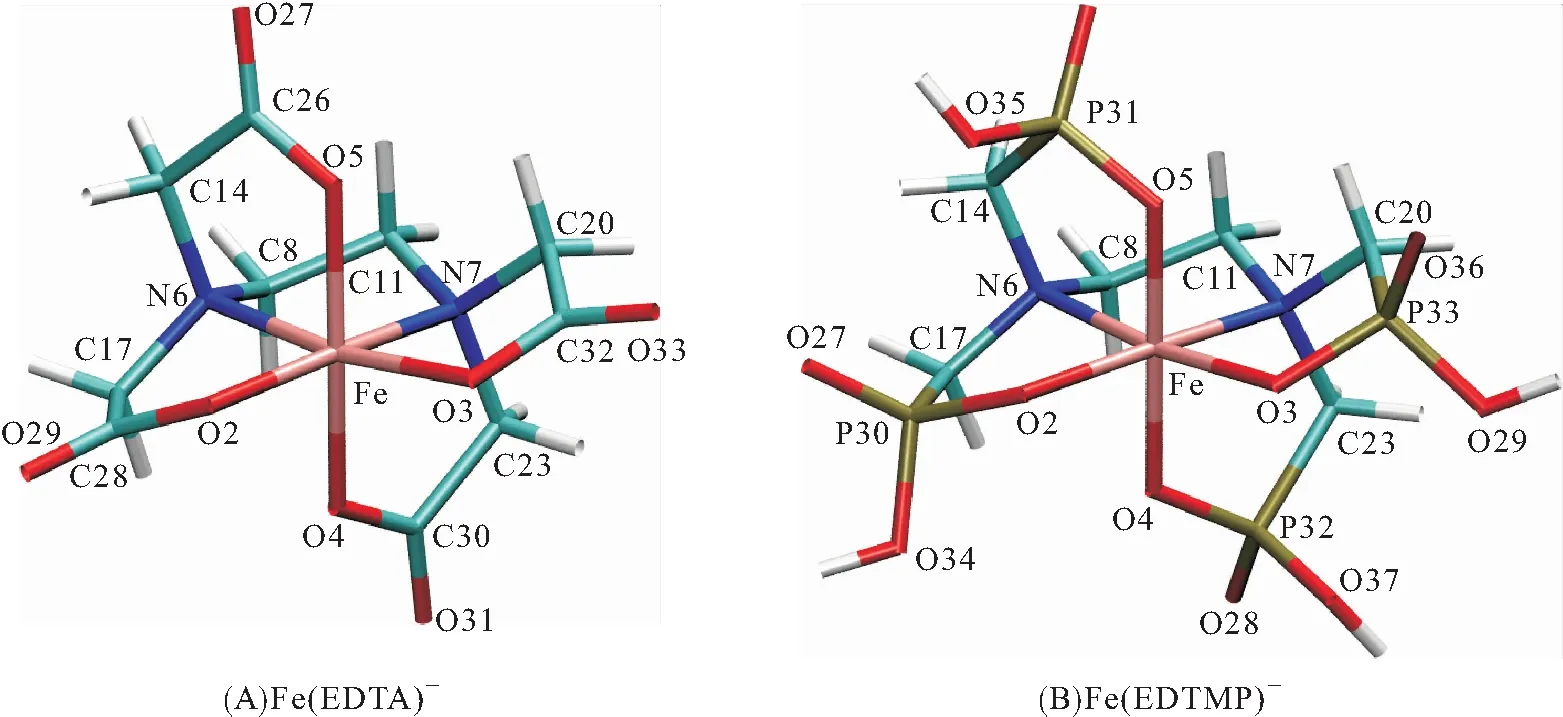
图1 配合物的优化构型

表2 低自旋态配合物的几何结构参数和Fuzzy键级Table 2 The geometry parameters and the Fuzzy bond orders ofthe low-spin complexes
EDTA和EDTMP的分子结构相似,差异在于EDTA有4个羧基,而EDTMP有4个膦酸基. 从表3中可以看出,由于C原子的电负性(2.55)比P原子的电负性(2.19)大,通过对电荷进行自然布局分析(NPA),EDTMP的膦酸基配位O原子的负电荷比EDTA的羧基配位O原子的负电荷多. 从单个基团的电荷分析,EDTMP上配位O原子的给电子能力应该比EDTA上配位O原子的更强,但实际效果并非如此,如果分析导致EDTMP溶解铁垢能力不如EDTA的强的因素,单方面用电荷多少判断结合力大小来分析并不合理,还需要考虑其他因素,例如空间位阻、拓扑结构、轨道相互作用等.

表3 配合物中部分原子的NPA电荷Table 3 The partial NPA charges of the complexes e
2.3 配合物的空间位阻分析
约化密度梯度是一种可视化研究弱相互作用的方法[13],不仅可反映范德华力、氢键和静电等弱相互作用,还能显示空间位阻作用. 弱相互作用临界点的电子密度ρ(r)是衡量相互作用强度的重要指标之一,其数值大小和键的强度存在正相关性,可用sign(2)函数表示键的类型. 键临界点的sign(2)=-1,环、笼临界点的sign(2)=+1. 将电子密度ρ(r)和sign(2)的乘积投影到RDG等值面上,可以可视化研究弱相互作用的强度与类型. 2种配合物分子的RDG等值面和RDG散点图如图2所示.

图2 配合物的RDG等值面和散点图
RDG等值面和散点图的颜色一致,图中蓝色区域代表起吸引作用的弱相互作用,绿色区域代表相互作用强度弱的范德华力;由于该区域电子密度很小,所以可正可负,其中绿色区域代表弱吸引作用、棕色区域代表弱互斥作用. 红色区域代表环、笼中原子间的互斥作用.
从图2中可以看出,2种配合物五元环内都有红色区域,表现为互斥作用. 2种分子的亚甲基上H原子和配位O原子间有绿色的弱相互吸引作用. 但2种配合物的配位基团之间相互作用并不相同,Fe(EDTMP)-的棕色区域明显大于Fe(EDTA)-的,表明Fe(EDTMP)-中相邻的膦酸基之间表现为弱互斥作用. 这是由于EDTA中羧基C原子为sp2杂化,EDTMP中膦酸基P原子为sp3杂化,sp3杂化比sp2杂化具有更大的空间伸展,导致Fe(EDTMP)-的膦酸基之间表现为弱位阻互斥作用,而Fe(EDTA)-的羧基之间位阻互斥作用更弱.
散点图中X、Y轴分别为sign(2)ρ(r)和RDG函数,将sign(2)ρ(r)范围定义为[-0.050~0.050],其中,sign(2)ρ(r)在[-0.025~-0.015]范围内表示强吸引,在[-0.015~0.015]范围内表示范德华力作用,在[0.015~0.025]范围内表示空间位阻作用. 从图2可以看出,在五元环中间有红色梭形区域,体现较强的位阻效应,对应于散点图最右边的红色峰. Fe(EDTMP)-在散点图[-0.015~0.015]范围内存在多个峰值,棕色区域体现膦酸基之间的弱位阻互斥作用,对应于[0.000~0.015]之间的峰;绿色区域体现配位O原子和亚甲基H原子之间存在微弱的范德华力,对应于[-0.015~0]之间的峰.
2.4 配合物的拓扑分析
BADER等[14]提出分子中的原子(Atoms in Molecules,AIM)理论,将化学结构、化学键等与电子密度分布函数的拓扑性质联系起来,定量描述了分子中原子及原子间化学键. 图3给出了2种配合物的拓扑分析图. 图中紫色点是(3,-3),对应于核临界点,与原子核的位置基本一致;桔色点是(3,-1),对应于键临界点(Bond Critical Point,BCP),位于2个有作用力的原子之间;黄色是环临界点. AIM理论认为,键临界点处的电子密度和能量密度与化学键性质存在密切联系. 表4列出2种配合物的配位键、1个羧基和1个膦酸基的键临界点性质.

图3 配合物的拓扑分析图

表4 配合物的键临界点的电子密度拓扑性质Table 4 The topological properties of bond critical points of the complexes
键临界点位置的电子密度ρ(r)和势能密度V(r)与化学键强度有密切关系. 对于同类化学键,通常ρ(r)越大、V(r)越负,则化学键强度越大. 由表4可知,Fe(EDTA)-中配位的4个O原子和2个N原子的ρ(r)均比Fe(EDTMP)-中的大,其V(r)则均比Fe(EDTMP)-的更负,说明Fe(EDTA)-的同类配位键强度均大于Fe(EDTMP)-的情况. 羧基键临界点的ρ(r)明显比膦酸基键临界点的大,这是由于羧基的OCO形成了离域π键,而膦酸基的P—O键为σ键. 羧基的离域程度大,影响了配位键临界点的电子密度,Fe(EDTA)-中键临界点的ρ(r)比Fe(EDTMP)-同类型键的大. 能量密度H(r)体现了某个点的电子能量,是动能密度G(r)与势能密度V(r)之和. 当H(r)<0时,化学键是共价作用,而当H(r)>0时,化学键是非共价作用[15].H(r)/ρ(r)的物理意义是BCP位置上单位电子的能量密度,H(r)/ρ(r)越负则共价作用越强,而H(r)/ρ(r)越正则非共价作用越弱[16]. 羧基比膦酸基的配位能力更强(表4),2种配合物的配位键临界点的H(r)和H(r)/ρ(r)均为负值,说明配位键为共价作用,且Fe(EDTA)-的配位键临界点的H(r)/ρ(r)均比Fe(EDTMP)-的更负,说明Fe(EDTA)-配位键的共价作用更强. 在2种配合物中,Fe—N配位键的H(r)/ρ(r)均比Fe—O配位键的更负,说明Fe—N配位键的共价作用强于Fe—O配位键. 这是因为N原子的电负性小于O原子,N原子束缚电子的能力小于O原子,其电子云具有更大的延伸性,与Fe3+形成共价键的共价作用更强.
2.5 配合物的自然键轨道分析
使用NBO程序对配合物进行自然键轨道(Natural Bond Orbital,NBO)分析. 表5列出部分电子供体(Donor)轨道i、电子受体(Acceptor)轨道j以及由二阶微扰理论得到轨道之间相互作用的稳定化能E(2).E(2)越大表明它们之间的相互作用越强,即轨道i供电子给轨道j的倾向越大,电子的离域化程度越大[17].

表5 配合物的主要电子供体和受体轨道的E(2) Table 5 The E(2) values of the main electron donor and acceptor orbits of the complexes
2种配合物分子的Fe—N反键轨道与其对面Fe—O反键轨道有最大离域作用,与其它Fe—O配位键的反键轨道也有离域作用,且Fe(EDTA)-的同类型E(2)均比Fe(EDTMP)-的大(表5). 羧基上O孤对电子与C—O反键轨道离域作用,明显强于膦酸基上O孤对电子与P—O反键轨道的相互作用,这与拓扑分析的结果一致. 图4展示了上述有强相互作用的轨道相互作用图,NBO轨道之间发生了重叠,使其轨道能量降低,这是其轨道稳定化能E(2)较大的原因. 拓扑分析和NBO分析均表明,EDTA中羧基形成离域π键,使得金属离子与配位原子间电子离域化程度变大,金属离子与配体共价作用增强,配位键强度增大. EDTMP由于膦酸基没有离域π键,不利于配位键共价作用的增强.
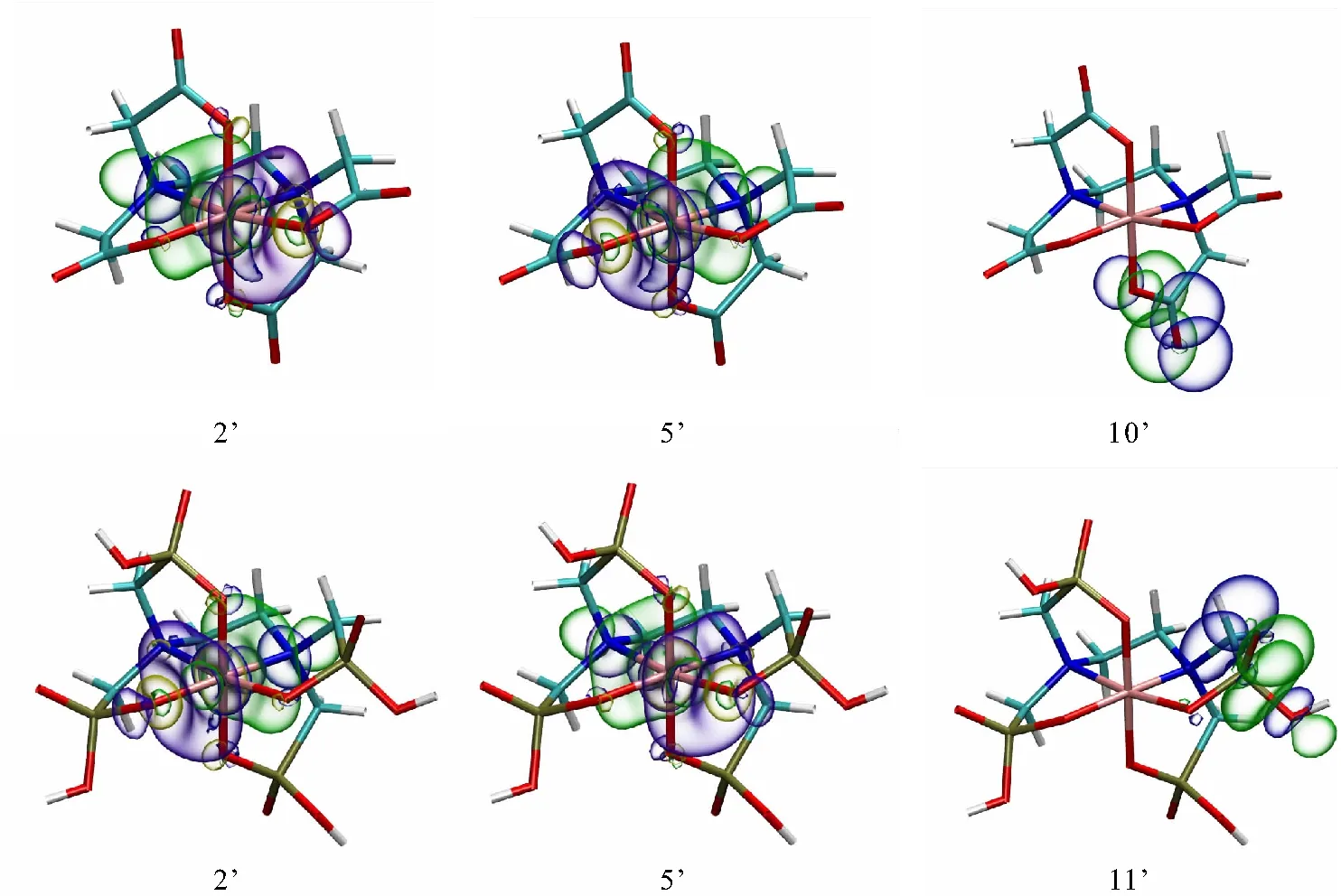
图4 配合物NBO轨道的相互作用图
3 结论
探讨了在铁垢的化学清洗过程中,只有EDTA可以溶解铁垢,而EDTMP不能够溶解铁垢的原因. 通过量子化学计算研究了两种配合物Fe(EDTA)-和Fe(EDTMP)-的分子结构,配合物为低自旋态的六配位八面体结构. 通过配合物分子的几何构型、电荷分布、扩展电荷分解分析、约化密度梯度分析、拓扑分析、自然键轨道分析发现,EDTA的羧基形成的离域π键,加强了配位键的相互作用,而EDTMP的膦酸基只有σ键. Fe(EDTA)-中配位键的共价作用强于Fe(EDTMP)-. EDTMP膦酸基之间的空间位阻不利于EDTMP与Fe3+结合. 结构上的差异导致EDTA与Fe3+的结合能力强于EDTMP. 本文从分子结构层面解释了EDTA常用于化学清洗,而EDTMP不能用于化学清洗的原因,为下一步的化学清洗分子设计奠定了基础.
猜你喜欢
橡塑技术与装备(2022年8期)2022-12-17
中国农业科学(2022年17期)2022-09-19
物理通报(2020年7期)2020-07-01
安徽化工(2018年3期)2018-07-04
科技创新导报(2017年19期)2017-09-13
政工学刊(2017年2期)2017-02-20
湖南师范大学学报·自然科学版(2014年4期)2014-10-23
中学生数理化·教与学(2008年3期)2008-07-11
