
Quantitation of DNA by nuclease P1 digestion and UPLC-MS/MS to assess binding efficiency of pyrrolobenzodiazepine
2020-07-02 01:59YongMaBuyunChenDongluZhang
Yong Ma,Buyun Chen,Donglu Zhang
Drug Metabolism and Disposition,Genentech,1 DNA Way,South San Francisco,CA,94080,USA
Keywords:
Nuclease P1
UPLC-MS/MS
DNA quantitation
DNA alkylation
Pyrrolobenzodiazepine(PBD-Dimer)
ABSTRACT
Accurate DNA quantitation is a prerequisite in many biomedical and pharmaceutical studies.Here we established a new DNA quantitation method by nuclease P1 digestion and UPLC-MS/MS analysis.DNA fragments can be efficiently hydrolyzed to single deoxyribonucleotides by nuclease P1 in a short time.The decent stabilities of all the four deoxyribonucleotides were confirmed under different conditions.Deoxyadenosine monophosphate(dAMP)was selected as the surrogate for DNA quantitation because dAMP showed the highest sensitivity among the four deoxyribonucleotides in the UPLC-MS/MS analysis.The linear range in DNA quantitation by this method is 1.2-5000 ng/mL.In the validation,the inter-day and intra-day accuracies were within 90%-110%,and the inter-day and intra-day precision were acceptable(RSD<10%).The validated method was successfully applied to quantitate DNA isolated from tumors and organs of a mouse xenograft model.Compared to the quantitation methods using UV absorbance,the reported method provides an enhanced sensitivity,and it allows for the accurate quantitation of isolated DNA with contamination of RNA and ribonucleotide.
1.Introduction
As the carrier of genetic information,DNA is essentially involved in a lot of modern chemical,biological and pharmacological studies.The methods of DNA isolation from different organisms have been maturely developed,and various commercial kits are available to provide a convenient and productive DNA isolation.In general,accurate quantitation of isolated DNA is not always necessary before the sequential step of characterization or application.For example,polymerase chain reaction(PCR)and PCR-based DNA sequencing usually characterize DNA qualitatively or only semiquantitatively.Meanwhile,DNA itself is also a therapeutic target of drugs including intercalating agents,alkylating agents,DNA cutters and many more[1].The inhibition of DNA replication can serve as an efficacious therapeutic strategy in the treatment of multiple diseases including cancers and virus infections.Pyrrolobenzodiazepine(PBD-dimer)is a DNA minor groove binder that forms covalent DNA interstr and cross-links in a sequencedependent manner(Fig.1)and exhibits broad-spectrum subnanomolar antiproliferative activities against a variety of cancer cell lines[2].In the studies involving DNA alkylating agents like PBD-dimer,accurate quantitation of DNA becomes necessary in evaluating DNA adduct occurrence[2].Conventionally,the concentrations of DNA solutions can be roughly determined by UV absorbance[3].Although the integrity of DNA fragments is kept by the UV absorbance method,the limitations of this method are also prominent:its low sensitivity and lack of robustness[4].The UV absorbance method usually tends to overestimate DNA concentrations,especially when isolated DNA is contaminated by RNA or ribonucleotides[5,6].Fluorescent dyes can help determine low DNA concentrations,but the sensitivity and accuracy are impacted by the various binding affinities between dyes and DNA fragments[7].

Fig.1.The scheme of DNA alkylkation by pyrrolobenzodiazepine(PBD-dimer).
DNA adducts are usually released via DNA hydrolysis,making the post-hydrolysis quantitation of DNA feasible.The method can be easily integrated with the steps of DNA adducts isolation and quantitation[2].DNA quantitation methods by hydrolysis,either chemically or enzymatically,have been introduced previously.Chemical hydrolysis of DNA usually requires harsh conditions which may change the structure of DNA adducts.Therefore,enzymatic digestion of DNA under physiological conditions is preferred in some cases.After digestion,DNA hydrolysis products can be analyzed by high performance liquid chromatography(HPLC)coupled with a UV detector.For example,Shimelis et al.[4]and Li et al.[8]established a nuclease P1 digestion/HPLC-UV method to quantitate DNA,and the DNA concentrations determined by this method were almost identical to those determined by the acid hydrolysis/HPLC-UV method.The hydrolysis/HPLC-UV methods were also found to be more reliable than non-hydrolysis methods including the dye-binding or direct UV spectrophotometric assays[4].Herein,by coupling tandem mass spectrometer(MS/MS)with nuclease P1 hydrolysis and reversed-phase UPLC,we developed a sensitive and efficient method to determine DNA concentration.The method has been fully validated and demonstrated to be useful in the quantitation of DNA isolated from tumors and organs in a mouse xenograft model,and it has been successfully applied to assess the DNA-alkylating efficiency of the PBD-dimer.
2.Experimental
2.1.Chemicals and reagents
Calf thymus(CT)DNA was purchased from Rockland Immunochemicals(Pottstown,PA,USA).Deoxyadenosine monophosphate(dAMP),thymidine monophosphate(TMP),deoxycytidine monophosphate(dCMP),deoxyguanosine monophosphate(dGMP),adenosine monophosphate(AMP),uridine monophosphate(UMP),cytidine monophosphate (CMP),guanosine monophosphate(GMP),nuclease P1 and deoxyribonuclease I(DNase I)were purchased from Sigma-Aldrich(St.Louis,MO,USA).Inosine monophosphate(IMP)was purchased from Cayman Chemical(Ann Arbor,MI,USA).DNeasy Blood & Tissue Kit was purchased from Qiagen(Valencia,CA,USA).Acetonitrile and water(MS grade)were purchased from EMD(Gibbstown,NJ,USA).
2.2.DNA digestion efficiency by enzymes
The DNA digestion by DNase I or nuclease P1 was initially characterized to determine whether they can digest DNA polymer to single deoxyribonucleotides.To test the DNA digestion efficiency of DNase I,10μg/mL CT DNA was incubated with 300 unit/mL DNase I for 2 h.The DNA digestion efficiency of nuclease P1 was further investigated to determine the amount of enzyme used and incubation time length.190μL of CT DNA solution(10 or 100μg/mL)was mixed with different concentrations of nuclease P1 in 10μL of water(0.1,0.01 or 0.001 unit)and then incubated at 37℃ for different time lengths(0,1,2,3,and 4 h).After digestion,the samples were diluted 200-fold in water.An aliquot of 200μL diluted samples was mixed with 50μL IMP in water(50 ng/mL)as the internal standard(IS)before UPLC-MS/MS analysis.
2.3.UPLC-MS/MS quantitation
The deoxyribonucleotides produced from DNA hydrolysis were analyzed on a Shimadzu Nexera HPLC system(Columbia,MD,USA)coupled to a Sciex API 6500 triple quadrupole mass spectrometer with an IonDrive Turbo V source(Sciex,Foster City,CA,USA)in positive ion mode.The conditions were as follows:column,Phenomenex XB-C18(100 mm × 2.1 mm,2.6μm);mobile phase A,water with 0.1%formic acid;mobile phase B,acetonitrile with 0.1%formic acid;gradient,0-1.0 min,0%B,1.0-2.0 min,0%-5%B,2.0-2.5 min,5%-95%B,2.5-3.0 min,95%B,3.0-3.5 min,95%-0%B,3.5-4.0 min,0%B; flow rate,1.0 mL/min;column temperature,50℃;and injection volume,10 μL.
The deoxyribonucleotides were quantitated in the multiple reaction monitoring(MRM)scan in the positive mode.The compound-dependent parameters are listed in Table 1,and the main instrument-dependent parameters were set as follows:ionspray voltage,5500 V;ion source temperature,500℃;collision gas(CAD),-3;curtain gas(CUR),30;nebulizer gas(GS1),60;and turbo gas(GS2),60.
2.4.Method validation
2.4.1.Stability and sensitivity of deoxyribonucleotides
Stability of the four deoxyribonucleotides in water was determined by analyzing QC samples(n=6)under different temperature conditions(at 90℃ for 30 min and at-80℃ for 14 days)or through three freeze-thaw cycles(-80℃-25℃).A signal-to-noise ratio of at least 10:1 was used as the principle criterion to determine the lower limit of quantification(LLOQ).Among the four deoxyribonucleotides,one with adequate stability and best sensitivity in MS analysis will be selected as a surrogate in DNA quantitation.
2.4.2.Calibration curve of DNA
Different concentrations of CT DNA in water were prepared as the standard samples to establish a calibration curve.After digestion by nuclease P1 and UPLC-MS/MS analysis,the peak area ratios of dAMP to IS were calculated and correlated with CT DNA concentrations by a least-squares linear regression method with 1/x2weighting(R2>0.9995).
2.4.3.Precision and accuracy
In the method validation,the precision and accuracy wasevaluated by quantitating the QC samples prepared at the concentrations(n=6 for each concentration).The inter-day and intraday precision and accuracy were calculated from the results of QC sample quantification on the same day and on three different days,respectively.

Table 1 Compound-dependent parameters in MS/MS analysis of mono-deoxyribonucleotides.
2.5.DNA extraction from tumors and organs of xenograft mice
The establishment of subcutaneous xenograft mouse model,administration of an anti-CD22 THIOMAB™antibody pyrrolobenzodiazepine (PBD)-dimerconjugate (with a cyclobutylcontaining disulfide linker),and sample collection were described previously[2,9].The tumor,liver,lung and kidney were weighed and then homogenized in 4-fold weight of ice-cold PBS.DNA from 75μL of homogenates was isolated by DNeasy Blood & Tissue Kit following instructions with modifications.After the first loading of tissue lysate,the flow-through was loaded two more times to ensure the best column binding of DNA.The columns were washed sequentially by two wash buffers(containing approximately 50%ethanol)provided in the kit.In the last step,DNA column was eluted with 200μL water twice and the elute was combined.
2.6.Release of PBD-dimer from DNA and quantitation
To digest DNA,0.001 unit of nuclease P1 in 10μL water was added to 190μL of mouse organ or tumor DNA sample and then incubated at 37℃ for 1 h.Digested samples were heated at 90℃ for 30 min to release the PBD-dimer.
The post-heating samples were aliquoted for the separate quantitation of DNA and PBD-dimer.For DNA quantitation,an aliquot of the post-heating samples was diluted in water by 200-fold.Before LC-MS/MS injection,200μL of diluted sample was mixed with 50μL of 50 ng/mL IMP in water as the IS.CT DNA was dissolved in water at various concentrations between 5 and 5000 ng/mL to serve as the standard curve for the DNA quantitation.For the quantitation of PBD-dimer,a standard curve of PBD-dimer can be made in either pure water or 100μg/mL CT solution.The standard curve samples made in CT DNA were incubated at 37℃ for 1 h before digestion and heating to ensure completion of DNA alkylation[2,9].It was observed that two standard curves in water and DNA solutions superimposed each other very well,indicating that the presence of deoxyribonucleotides did not impact the recovery and quantitation of PBD-dimer.Also,PBD-dimer was confirmed to be stable in the absence of DNA,as well as at high temperature(90℃).Thus,quantitative recovery of PBD-dimer from tissue DNA samples can be expected after the digestion and heating process.The LC-MS/MS method for PBD-dimer has been described previously[2,9].
The calculation of PBD-DNA adducts abundance is based on the results of DNA and PBD-dimer quantitation.The mass of DNA was converted to the number of base pairs,based on an average DNA base pair molecular weight as 650 Da.The final results were shown as the adduct numbers per 106base pairs.
2.7.DNA quantitation by UV
The absorbance readings of DNA or DNA/nucleotides samples at different wavelengths(230,260,and 280 nm)were determined by a NanoDrop 8000 spectrophotometer(NanoDrop Technologies Inc.,Wilmington,DE,USA).The DNA concentrations were calculated from the absorbance reading at 260 nm(1 OD260 unit=50μg/mL pure DNA).
3.Results and discussion
3.1.Chromatography and mass spectrometry
MRM transitions and parameters for the quantitation of deoxyribonucleotides were determined by manual tuning(Table 1).For a better ionization in the source,0.1%formic acid in water and 0.1%formic acid in acetonitrilewere used as the mobile phase A and B,respectively.Multiple reversed-phase columns were tested and Phenomenex XB-C18column(100 mm × 2.1 mm,2.6μm)was selected to provide the best retention and peak shape.To improve the peak shape of hydrophilic analytes,the column temperature was set as 50℃ and the flow rate was set as 1 mL/min.In the liquid chromatography,the retention times of dAMP,TMP,dCMP and dGMP were 1.41,0.98,0.54 and 1.22 min,respectively.IMP,a ribonucleotide with a similar structure,was selected as the internal standard in the quantitation.The retention time of IMP is 0.80 min(Fig.2A).
3.2.The DNA digestion efficiency of nuclease P1
Incubation with nuclease P1 completely digested CT DNA to single deoxyribonucleotides(Fig.2B).To figure out whether nuclease P1 can be substituted by another commonly used DNA-cutting enzyme,DNase I,the cleavage of CT DNA by DNase I was also tested.The result showed that no single deoxyribonucleotides were released,indicating that DNase I can only cut DNA into smaller fragments or oligos(Fig.2E).
The DNA digestion efficiency of nuclease P1 was investigated to determine the amount of nuclease P1 needed to digest certain unit weight of DNA and the appropriate incubation time as well.Relatively high concentration DNA solutions(10 or 100μg/mL)was digested by different concentrations of nuclease P1(final concentrations as 0.5,0.05 and 0.005 unit/mL)for various periods of time(0,1,2,3 and 4 h).Based on the peak areas of deoxyribonucleotides,it was observed that the digestion of DNA by nuclease P1 was completed very quickly in all the incubations(Fig.3).With the lowest enzyme concentration tested(0.005 unit/mL),the digestion of 100 μg/mL DNA was finished within 1 h of incubation at 37℃.With 0.5 or 0.05 unit/mL nuclease P1,the digestion was almost 100%completed even before the 37℃ incubation began,indicating the great efficiency of nuclease P1 in DNA digestion.Therefore,0.005 unit/mL nuclease P1 and 1 h,37℃ incubation were selected as the conditions for DNA digestion.
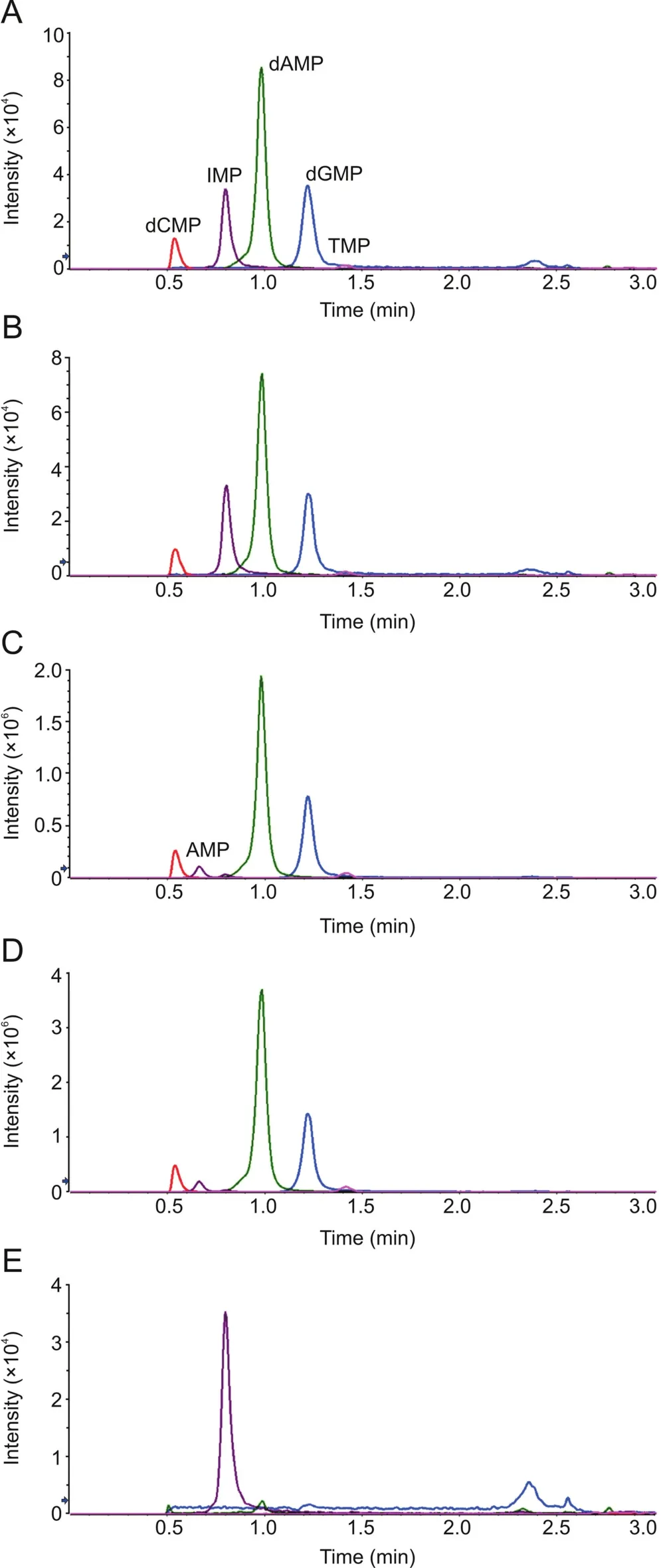
Fig.2.Representative chromatograms in UPLC-MSMS analysis:(A)20 ng/mL equimolar mixture of the 4 deoxyribonucleotides;(B)20 ng/mL CT DNA after nuclease P1 digestion;(C)100 μg/mL CT DNA spiked with 100 μg/mL ribonucleotide equimolar mixture,after nuclease P1 digestion(200-fold diluted before injection);(D)DNA isolated from mouse tumor,after nuclease P1 digestion(200-fold diluted before injection);(E)20 ng/mL CT DNA after DNase I digestion.
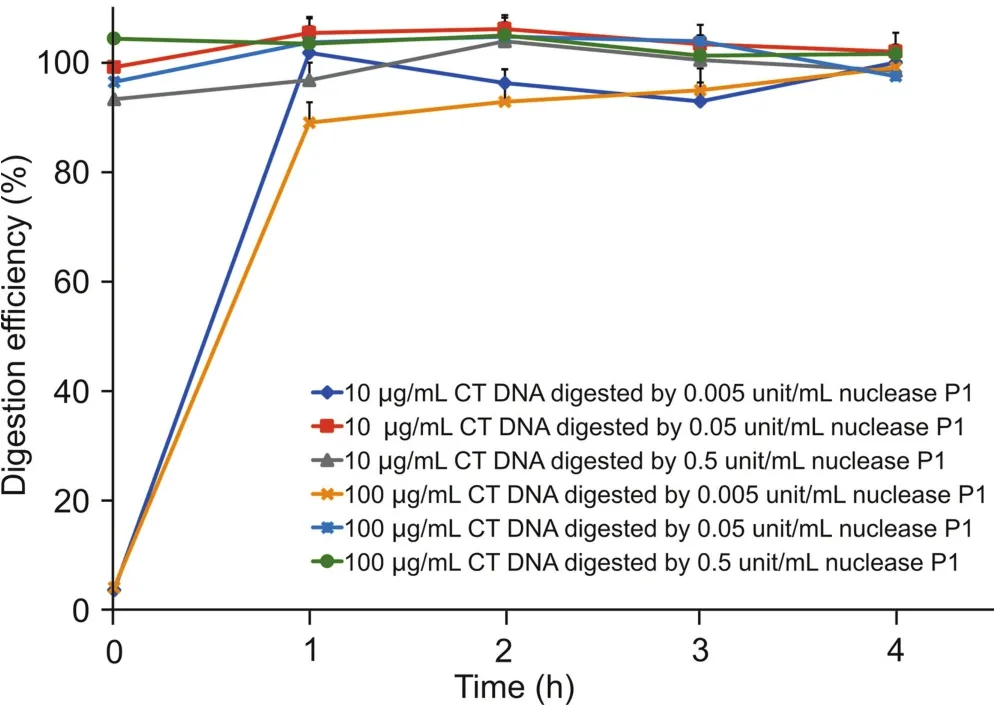
Fig.3.The time course of calf thymus DNA digestion by nuclease P1.10 or 100μg/mL calf thymus DNA was incubated with 0.005-0.5 units/mL nuclease P1 for up to 4 h to determine the digestion efficiency.
3.3.Method validation
3.3.1.Stability of deoxyribonucleotides
The stability of deoxyribonucleotides was investigated under different conditions,including at high temperature for a short time(90℃ for 30 min),low temperature for long-term storage(-80℃ for 14 days),and undergoing repeated freeze-thawaction(3 cycles).These conditions were selected to assess the potential impact of practical operations on post-digestion DNA quantitation.In all the tests,the four deoxyribonucleotides exhibited excellent stability,with almost 100%remaining after all the treatments(Table 2).The con firmed stability of dAMP,TMP,dCMP,and dGMP indicated that deoxyribonucleotides derived from DNA digestion can serve as solid surrogates in DNA.
3.3.2.Sensitivity and linearity
Using authentic deoxyribonucleotides as standards,the LC-MS/MS method was found to be with various sensitivities in detecting each of them.The LLOQs for dAMP,TMP,dCMP,and dGMP were determined as 0.3,4.8,1.2 and 1.2 ng/mL,respectively.Therefore,dAMP was selected as the surrogate after DNA hydrolysis by nuclease P1,and the linear range for DNA quantitation was determined as 1.2-5000 ng/mL.DNA isolated from cell or tissue samples with a concentration higher than 5000 ng/mL should be appropriately diluted before quantitation.
3.3.3.Accuracy and precision
Six replicate CT DNA samples at three concentrations(10,100,and 1000 ng/mL)as QC were measured to determine the intra-day and inter-day accuracy and precision.The results shown in Table 3 indicate that for both intra-day and inter-day,the accuracies were always between 90%-110%and the precisions were within the acceptable range(relative standard deviation lower than 10%).
3.4.The quantitation of DNA isolated from tissue samples
In addition to the enhanced sensitivity,the superiority of DNA quantitation by nuclease P1 digestion and UPLC-MS/MS over thetraditional UV absorbance method was further examined.DNA isolated by commercial kits is often contaminated by RNA unless the samples are pre-treated with RNase before extraction[10,11].In the presence of RNA or RNA-derived ribonucleotides,a higher UV absorbance can be expected and it will lead to an overestimation of DNA concentrations.For example,after samples containing known concentrations of CT DNA were spiked with ribonucleotides,the accurate DNA concentration could not be determined by the UV absorbance method(Table 4).The direct quantitation of DNA by UV absorbance relies on both relatively high DNA concentration and good DNA purity.The HPLC-UV method may exclude the inferences from RNA or ribonucleotides by separating the peaks in the chromatography,which takes more efforts in the LC method development.Quantitation of DNA by digestion and LC-MS/MS is not limited by the purity of DNA samples.CT DNA was still accurately quantitated by this method after spiked with ribonucleotides(Fig. 2C), indicating that the signal channels of deoxyribonucleotides in mass spectrometry were not impacted by the presence of RNA or ribonucleotides(Table 4).
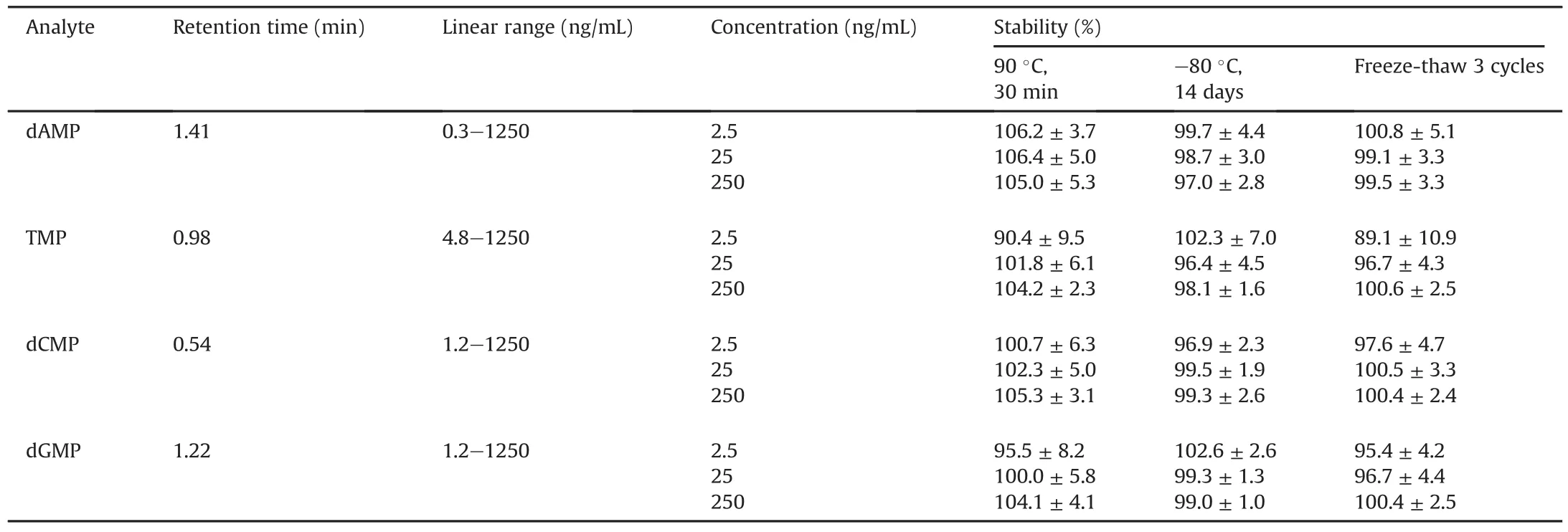
Table 2 LC retention times,LC-MS linear ranges,and stabilities of deoxyribonucleotides.

Table 3 Method validation for LC-MS/MS analysis of DNA.
3.5.The PBD-DNA abundance in the mouse tumors and organs
Previously,we reported the in vitro and in vivo characterization of antibody-drug conjugates(ADCs)bearing a PBD-dimer as the cytotoxic payload[2,9].To fully assess the anti-tumor efficacy of the ADCs in a mouse xenograft model,the abundance of DNA-PBDadducts in tumor or organs was investigated.As shown in Table 4,the DNA concentrations isolated from mouse tumors and tissues were all overvalued by UV absorbance in varying degrees,probably due tothe contamination of RNA or ribonucleotides.In contrast,the digestion and UPLC-MS/MS method excluded the interferences from RNA or ribonucleotides(Fig.2D).In addition,this method can be integrated with UPLC-MS/MS quantitation of DNA alkylators or adducts,providing a convenient and comprehensive way to determine the DNA adduct occurrence.By employing this method in our studies,the accurate numbers of DNA adducts formed per million base pairs have been successfully determined and correlated with the anti-tumor efficacy of the ADCs(Table 5).

Table 4 Comparative quantitation of DNA by UV and LC-MS/MS of dAMP after DNA hydrolytic digestion.

Table 5 DNA isolation and PBD-dimer recovery in tumor and major organs from xenograft mice after the administration of an anti-CD22 THIOMAB™antibody PBD-dimer conjugate(with a cyclobutyl-containing disulfide linker).
4.Conclusions
Digestion by nuclease P1 can efficiently convert DNA fragments to single deoxyribonucleotides. The stability of deoxyribonucleotides was confirmed in various conditions and the results demonstrated that they can serve as the surrogate of DNA for the quantitation purpose.Here,DNA digestion and UPLC-MS/MS analysis of post-digestion deoxyribonucleotides constitute an accurate,sensitive and convenient method to quantitate DNA.More importantly,this method can exclude the interferences by RNA or ribonucleotides when DNA samples contain such impurities.The analytical method we described here can be used for the accurate DNA quantitation,which can be crucial for many downstream experiments to study DNA.Since DNA is an important drug target in oncology,this method can be highly useful in determining the DNA adduct abundance in tumor cells and help understand the mechanism of action of DNA alkylating agents(like PBD-dimer).
Conflicts of interest
The authors declare that there are no conflicts of interest.
 Journal of Pharmaceutical Analysis2020年3期
Journal of Pharmaceutical Analysis2020年3期
- Journal of Pharmaceutical Analysis的其它文章
- Current status and future directions of high-throughput ADME screening in drug discovery
- Current LC-MS-based strategies for characterization and quantification of antibody-drug conjugates
- Current status of in vivo bioanalysis of nano drug delivery systems
- Ultra-sensitive bioanalysis of the therapeutic peptide exenatide for accurate pharmacokinetic analyses at effective plasma concentrations utilizing UPLC-MS/MS
- Software-aided detection and structural characterization of cyclic peptide metabolites in biological matrix by high-resolution mass spectrometry
- Rapid bioluminescence assay for monitoring rat CES1 activity and its alteration by traditional Chinese medicines