
Histopathological landscape of rare oesophageal neoplasms
2020-08-24 07:29GianlucaBusinelloCarloAlbertoDalPozzoMartaSbaragliaLucaMastracciMassimoMilioneLucaSaragoniFedericaGrilloPaolaParenteAndreaRemoElenaBellanRoccoCappellessoGianmariaPennelliMauroMichelottoMatteoFassan
World Journal of Gastroenterology 2020年27期
Gianluca Businello, Carlo Alberto Dal Pozzo, Marta Sbaraglia, Luca Mastracci, Massimo Milione, Luca Saragoni,Federica Grillo, Paola Parente, Andrea Remo, Elena Bellan, Rocco Cappellesso, Gianmaria Pennelli,Mauro Michelotto, Matteo Fassan
Abstract The landscape of neoplastic pathology of the oesophagus is dominated by malignancies of epithelial origin, in particular by oesophageal adenocarcinoma and oesophageal squamous cell carcinoma. However, several other histopathological variants can be distinguished, some associated with peculiar histopathological profiles and prognostic behaviours and frequently underrecognized in clinical practice. The aim of this review is to provide a comprehensive characterization of the main morphological and clinical features of these rare variants of oesophageal neoplastic lesions.
Key words: Gastrointestinal tumours; Oesophageal tumours; Histopathology; Rare tumours; Mesenchymal tumours
INTRODUCTION
The landscape of neoplastic pathology of the oesophagus is dominated by malignancies of epithelial origin, in particular, by oesophageal adenocarcinoma(OAC) and oesophageal squamous cell carcinoma (OSCC), which are by far the most common malignant neoplasms of the oesophagus. As the eighth most common cancer in the world, oesophageal cancer affects more than 450000 people/year worldwide,with a 5-year survival of 17% in metastatic cases[1,2]. Since the typical clinical presentation of oesophageal space-occupying lesions is worsening dysphagia (i.e.,“subjective sensation of difficulty in the swallowing of solids”), patients with this symptom are primarily studied in order to exclude the presence of OAC or OSCC.Nevertheless, several other neoplastic and non-neoplastic conditions can present with similar clinical symptoms, thus making the differential diagnosis more difficult.
The aim of this paper is to provide a comprehensive view of neoplastic histotypes occurring within the oesophageal wall that are rarer than OSCC and OAC (Figure 1 and 2). Biological features, clinicopathologic characteristics, major diagnostic issues and potentially useful diagnostic means will be discussed for each entity as evaluated by PubMed search results (April 2020). When available, the molecular background of the disease will be provided. Finally, a description of up-to-date therapeutic approaches adopted in patients with these diseases will be given together with relevant prognostic implications. Since all of these tumours are exceedingly rare, no robust data about each one’s incidence are available.
RARE PRIMARY OESOPHAGEAL BENIGN TUMOURS AND TUMOURS OF UNCERTAIN BEHAVIOUR
Primary benign tumours of the oesophagus are overall rare, accounting for 1% or less of all oesophageal tumours at autopsy. In this category, the most common tumour type is leiomyoma[3].
Oesophageal leiomyoma
Background:Oesophageal leiomyomas (OLMs) are the most common benign tumours of the oesophagus, accounting for 70%-80% of cases[3,4]. OLMs generally arise from the inner circular muscle layer of the distal and midthoracic portion of the oesophagus. Such tumours typically occur in middle-aged patients with a male predominance[5,6]. The coexistence of oesophageal and female genital leiomyomas is described as oesophagovulvar syndrome, of which only 14 cases have been reported since 1953[7].
Clinical features and diagnostic approach:In half of patients, OLMs are asymptomatic. Patients with symptomatic OLMs typically complain of dysphagia, epigastric pain and /or pyrosis. Symptoms most often appear when the tumour’s diameter exceeds the critical point of 4.5-5 cm[5,6]. The diagnostic approach includes oesophagogastroduodenoscopy (EGDS), endoscopic ultrasonography (EUS), magnetic resonance imaging (MRI) and oesophagogram, while the utility of preoperative biopsy is debated[4,6].
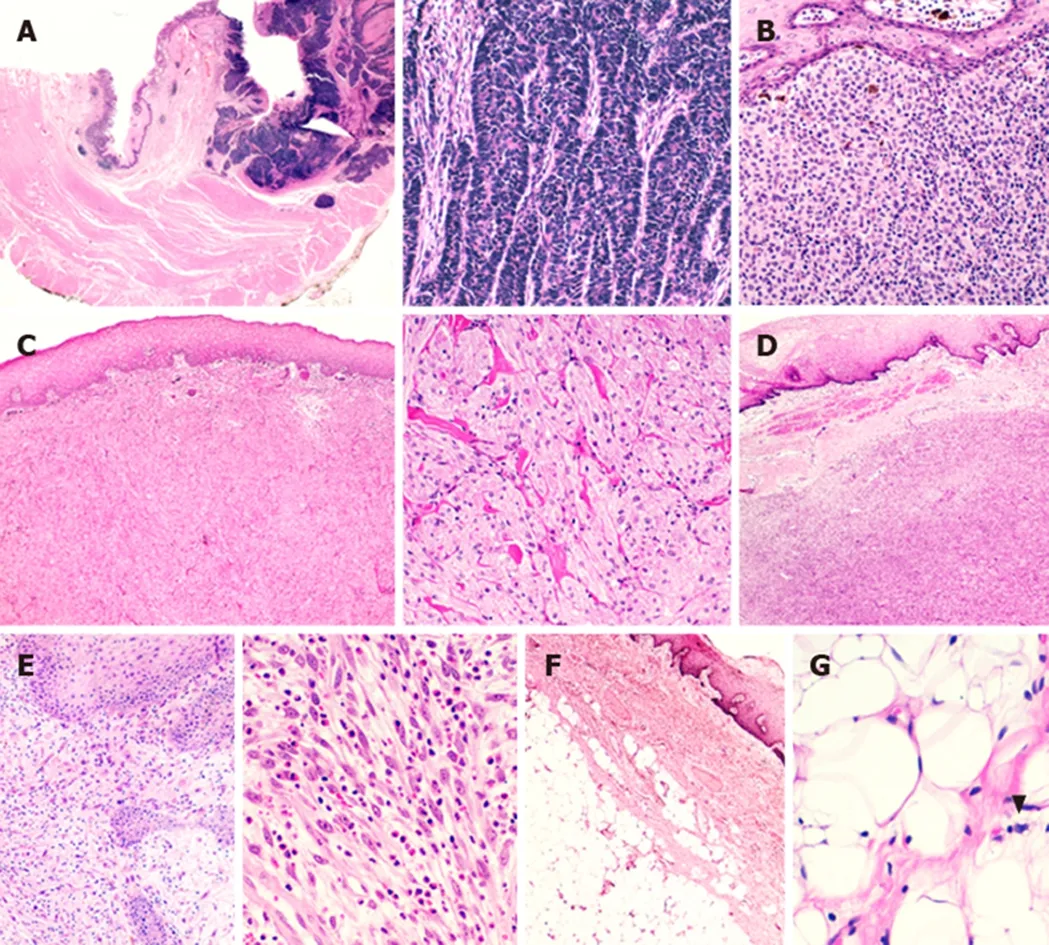
Figure 1 Representative rare histotypes of oesophageal neoplastic lesions. A: Neuroendocrine carcinoma with full-thickness oesophageal wall involvement; at higher magnification, note the organoid growth pattern of the lesion;B: A case of primitive oesophageal melanoma with melanin deposits; C: Granular cell tumour of the oesophageal wall; note the sheets or nests of plump, round or polygonal cells with eosinophilic granular cytoplasm at higher magnification; D: Oesophageal gastrointestinal stromal tumour. E: Inflammatory myofibroblastic tumour characterized by spindled myofibroblasts and ganglion-like cells dispersed in a myxoid background with a lymphocytic, plasma cellular and eosinophilic infiltrate. F: Oesophageal lipoma; G: High-power field of an atypical oesophageal lipomatous tumour (i.e., ”well-differentiated liposarcoma”) (arrowhead showing a lipoblast).
Pathological features:Microscopically, OLMs are composed of bundles of welldifferentiated spindle-shaped smooth muscle cells without clear demarcation or a well-formed capsule. Mitoses are not usually detected, but mild focal nuclear atypia can be present. On immunohistochemistry, leiomyomas are positive for desmin,smooth muscle actin (SMA), caldesmon and calponin[8]. Interestingly, OLMs can show numerous mast cells, interstitial cells of Cajal and expression of cKIT, which can give rise to a challenging differential diagnosis from gastrointestinal stromal tumours(GISTs).
Treatment:The location and size of the leiomyomas are important factors in the choice of the most appropriate surgical approach. In small (2-4 cm) tumours,endoscopic approaches such as endoscopic mucosal resection (EMR) and submucosal dissection (ESD) are possible, while in larger tumours, open surgery may be necessary[6]. The prognosis is excellent[5].
Oesophageal hamartoma
Background:Hamartomas are defined as tumour-like malformations composed of a mixture of differentiated normal tissues physiologically involved in the structure of the organ in which they develop. To date, fewer than ten reports describe an oesophageal location of these tumours[9].
Clinical features and diagnostic approach: Small oesophageal hamartomas (OHs)can be asymptomatic or can cause progressive dysphagia, often accompanied by weight loss due to diminished caloric intake. OHs are usually located in the submucosa; thus, a combination of endoscopic ultrasound, barium oesophagogram and contrast-enhanced computed tomography (CT) with classic EGDS and biopsy sampling[8]can be very helpful in patient work-up[9].
Pathological features:OHs are typically composed of a mixture of fat, fibrous, and mature cartilaginous tissues associated with benign glandular structures[9].
Treatment:Surgery is considered curative[9].
Oesophageal haemangioma
Background:Oesophageal haemangiomas (OHAs) are rare benign tumours, with fewer than a hundred cases reported in the literature. OHAs seem to have a first peak incidence in the fourth decade in both sexes and a second peak in the sixth decade in males[10].
Clinical features and diagnostic approach:Patients usually complain of dysphagia(45.2%), haematemesis (25.8%), melena (12.9%) and retrosternal pain (12.9%); a risk of severe haemorrhage does exist[10,11]. EUS and CT scans are useful for correct clinical assessment[10,11].
Pathological features:OHAs are commonly located in the mucosa or submucosa.They may have variable histological features, but the overarching morphologic characteristic is the presence of thin-walled, blood-filled vessels lined by a single layer of flat, cytologically banal endothelial cells that are usually positive for CD31, ERG,CD34 and FLI1 on immunohistochemistry[12].
Treatment:Endoscopic resection, when possible, is the gold standard procedure.Surgical resection and laser fulguration are also considered curative[10,11,13].
Oesophageal glomus tumour
Background:Glomus tumours are mesenchymal neoplasms composed of cells resembling the modified smooth muscle cells of the normal glomus body. They account for less than 2% of soft tissue tumours[12]. These neoplasms are usually found in subcutaneous tissues of distal extremities, but they can also occur in different anatomic sites. In particular, oesophageal glomus tumours (OGTs) are extremely rare,with fewer than ten cases described in the literature[14]. Despite generally being considered benign lesions, three cases of OGT with malignant behaviour have been reported in the literature[15-17].
Clinical features and diagnostic approach:Benign OGTs generally arise in the mucosal and/or submucosal layer. They may be asymptomatic or cause vague discomfort, pain and heat in the neck or dysphagia, whereas patients with malignant OGTs can additionally present with weight loss[14,18,19]. Familial glomus tumours show autosomal dominant inheritance caused by inactivating mutations in theGLMNgene or in association with neurofibromatosis type 1. To the knowledge of authors, OGTs have never been reported in inherited syndromes. Patients with malignant OGTs present with dysphagia and weight loss[12,15,17]. Due to the rarity of these lesions and the associated nonspecific symptoms, making a correct preoperative diagnosis is challenging. EUS and CT scans may be useful for the correct assessment of patients;however, the diagnosis is made after histological analysis of biopsy or surgical specimens[14].
Pathological features:Microscopically, OGT cells are distributed around the blood vessels and appear small, round and uniform, with a central, round nucleus and a granular eosinophilic cytoplasm. Cell borders are sharply defined, and the stroma may show hyalinization or myxoid change[12,14]. In malignant OGTs, necrosis, high mitotic activity and marked nuclear atypia have been found[15,17]. On immunohistochemistry, OGTs are positive for SMA, caldesmon and CD34[14,15,17].Collagen IV and laminin immunostaining can highlight the abundant pericellular basal membrane material. Occasionally, desmin and CD34 are focally positive.
Treatment:In benign OGTs, both endoscopic and surgical resection are considered curative. Malignant OGTs were treated with surgical resection alone, and the prognosis is reported to be good[15-17].
Oesophageal schwannoma
Background:Schwannomas are tumours of the peripheral nervous system that frequently occur in the head and neck region, extremities and retroperitoneum.Oesophageal schwannomas (OSs) are extremely rare, with just a few dozen cases reported in the literature[20-22]. OSs are most commonly observed in the submucosal layer of the upper oesophagus of middle-aged women[22].
Clinical features and diagnostic approach:Dysphagia is, by far, the most common symptom in patients with OSs[21]. EGDS, EUS and CT scans are useful for correct clinical assessment (Figure 2B)[20,21]. Some OSs have been reported in association with neurofibromatosis type 2[20].
Pathological features:OSs, and schwannomas of the gastrointestinal tract in general,have different characteristics compared to their soft tissue counterparts. OSs are unencapsulated but well-circumscribed tumours that are characterized by spindle cells with microtrabecular architecture. OSs can also present focal cellular pleomorphism with nuclear atypia, rare mitotic figures and a lymphoid infiltrate, also containing germinal centres[8,21]. Gastrointestinal schwannomas usually lack typical features present in soft tissue counterparts, such as encapsulation, nuclear palisading,vascular hyalinization and dilatation[8]. On immunohistochemistry, OSs are positive for S100 and SOX10 but negative for CD34, SMA, desmin and CD117[21].
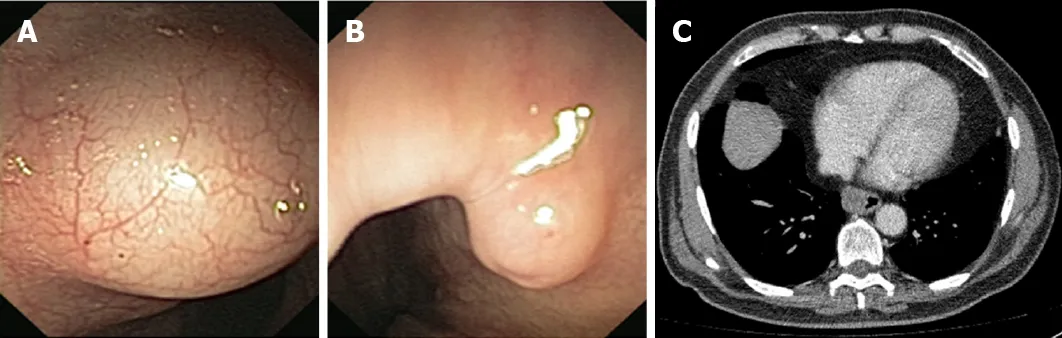
Figure 2 Representative endoscopic and radiologic features of rare histotypes of oesophageal neoplastic lesions. A: Endoscopic appearance of a submucosal oesophageal lipoma; B: A protuberant oesophageal schwannoma is evident at endoscopy; C: Computed tomography scan showing an oesophageal gastrointestinal stromal tumour (3.5 cm of diameter) located at the inferior third of the oesophageal wall.
Treatment:Surgical enucleation is considered curative[20-22].
Oesophageal calcifying fibrous tumour
Background:Calcifying fibrous tumours are rare benign fibroblastic tumours that affect both sexes equally and are more typical of children and young adults than older patients[23]. These neoplasms may develop in many different anatomic sites (gastric location being the most common), but oesophageal calcifying fibrous tumours(OCFTs) are extremely rare, with fewer than ten cases described in the medical literature[23-25].
Clinical features and diagnostic approach:Due to the possible different locations,calcifying fibrous tumours may cause a wide range of symptoms that are generally nonspecific. A subset can be multifocal and may be associated with other diseases,such as Castleman disease, inflammatory myofibroblastic tumour, vascular anomaly and sclerosing angiomatoid nodular transformation[24,25]. OCFTs described in the medical literature were generally clinically silent and discovered incidentally as a mass located in the deep mucosa, submucosa and/or the muscularis propria; one patient reported a foreign body sensation in the throat[22,24]. EUS, doppler flow imaging, contrast-enhanced CT scan and/or MRI can be helpful in the clinical assessment, but the final diagnosis of OCFT is made after histopathological evaluation[23-25].
Pathological features:OCFTs are characterized by an unencapsulated proliferation of spindle cells embedded in a hyalinized stroma with prominent chronic inflammation(i.e., plasma cells and small lymphocytes) and both dystrophic and psammomatous calcifications[23-25]. On immunohistochemistry, tumour cells are positive for CD34 and,in a minor subset, SMA. Immunohistochemistry for AE1/AE3, desmin, CD117, DOG-1, S100 and anaplastic lymphoma kinase 1 (ALK1) is negative. Increased amounts of IgG4 plasma cells have been reported. The Ki67 proliferation index is usually low (1-3%)[25].
Treatment:Surgery is considered curative[23-26].
Oesophageal lipoma
Background:Oesophageal lipomas (OLs) are particularly rare, accounting for less than 0.5% of all benign neoplasms of the digestive tract, and most commonly involve the submucosal layer of the proximal oesophagus[27,28].
Clinical features and diagnostic approach:OLs smaller than 1 cm are clinically silent,and only patients bearing large tumours present with dysphagia, regurgitation,recurrent melena, heartburn and/or a sense of “fullness” in the throat[28,29]. Notably,despite being proper benign neoplasms, OLs might become a life-threatening disease due to the risk of asphyxiation[28]. MRI is the preferred noninvasive technique for the diagnosis of OLs, even if EGDS with biopsy sampling may be required (Figure 2A).
Pathological features:OLs are described to feature a collection of mature adipose tissue (Figure 1F)[27].
Treatment:OLs measuring less than 1 cm in diameter are often asymptomatic and do not require treatment. Lesions with diameters of approximately 2-5 cm can be treated by endoscopic resection. Surgical resection is the first-line therapy for large (more than 5 cm) and symptomatic OLs[27,28].
Oesophageal lymphangioma
Background:Lymphangiomas (LAs) are benign soft tissue tumours that should actually be considered congenital malformations rather than neoplasms[30]. An oesophageal location of LAs is exceedingly rare, with only 22 cases reported worldwide[31]. Notably, even though the diagnosis of LAs is typically made in children younger than two years of age, all oesophageal cases have been described in the distal oesophagus in middle-aged men[31].
Clinical features and diagnostic approach:Oesophageal LAs smaller than 2 cm are clinically silent, while dysphagia is the chief symptom of patients bearing larger lesions. Oesophageal LAs are generally located in the submucosal layer, eventually involving the muscularis propria. The adequate diagnostic assessment requires the use of EUS examination of the oesophageal wall, which shows the cystic nature of LAs. When EUS findings are not typical, the definitive diagnosis of oesophageal LAs requires histological examination[31].
Pathological features:Oesophageal LAs are characterised by thin walled, dilated lymphatic channels lined by flattened endothelium, which can be empty or filled with chylous or serous fluid, with the occasional presence of lymphocytes or erythrocytes.Additionally, these tumours are frequently surrounded by lymphocytic aggregates[31-33].
Treatment:Small LAs (i.e., less than 2.5 cm in size) that leave the oesophageal muscles intact can be managed by ESD. Surgical treatment is needed when tumours are large and involve the muscular layer[31].
Oesophageal inflammatory fibroid polyps
Background:Inflammatory fibroid polyps are uncommon benign tumours that can arise along the gastrointestinal tract. The stomach is the most common location, while ten cases of oesophageal inflammatory fibroid polyps (OIFPs) have been reported in the medical literature[34,35].
Clinical features and diagnostic approach:OIFPs are usually located in the submucosal layer of the distal oesophagus. The most common symptom of OIFPs is dysphagia, but patients can also present with bleeding and gastro-oesophageal reflux disease. Occasionally, OIFPs can be asymptomatic[34,35]. EGDS with bioptic sampling is the most useful diagnostic tool; alternatively, patients can be investigated with CT scans and MRI[34,35].
Pathological features:Microscopically, OIFPs are characterized by proliferation of fibroblasts and small vessels in a myxoid stroma with plasma cells and a diffuse eosinophilic infiltrate[34,35]. On immunohistochemistry, positivity for CD34 and SMA has been described[35].
Treatment:Surgical resection is considered curative[34,35].
Oesophageal granular cell tumour
Background:Granular cell tumours (GCTs) (or granular cell myoblastomas or Abrikossoff tumours) are uncommon neoplasms showing neuroectodermal differentiation[36]. Oesophageal granular cell tumours (OGCTs) account for approximately one-third of all gastrointestinal GCTs. They are more typical of middle-aged females (male to female ratio 6:8) and are more often situated in the deep mucosa and/or submucosa of the distal segment of the oesophagus (65% of cases)[36,37].
Clinical features and diagnostic approach:OGCTs measuring less than 2 cm are clinically silent, whereas larger ones may cause dysphagia, gastroesophageal reflux disease, dyspepsia, chest pain, cough, nausea and hoarseness[36]. A critical prerogative of GCTs is their potential malignant degeneration (1%-2% of cases). In light of this, the diagnostic approach for these tumours should primarily be aimed at excluding the malignant behaviour of these lesions[38]. The most useful tool for preoperative diagnosis and assessment of tumour size, location and depth of invasion is EUS[37].Further aid in diagnostic assessment is provided by EGDS with biopsy sampling, CT scan and/or MRI. The typical endoscopic appearance of benign OGCTs is a yellowish,smooth surfaced, firm lesion covered by intact mucosa. Conversely, mucosal ulceration is suspicious for malignancy[36]. Multiple GCTs may arise in association with Noonan syndrome, although it has never been reported in the oesophagus.
Pathological features:The microscopic hallmarks of benign GCTs are sheets or nests of plump, round or polygonal cells with eosinophilic granular cytoplasm and uniform, small, round and pyknotic central nuclei (Figure 1C). The cells are often separated by collagen fibre bundles, and necrosis must be absent[36].Immunohistochemical staining is indispensable to demonstrate the expression of biomarkers attesting to the neural origin of GCTs, such as S100 protein[37]. Expression of these biomarkers, together with negative immunoreaction for smooth muscle actin and desmin, definitively rules out leiomyoma from the differential diagnoses (the most common submucosal lesion of the distal oesophagus)[36]. The pathological estimation of malignant potential is assessed by the conventional criteria proposed by Fanburg-Smith: Tumours fulfilling at least three features among (1) Necrosis; (2)Spindling, vesicular nuclei with large nucleoli; (3) High nuclear-to-cytoplasmic ratio;(4) Increased mitotic activity (i.e., more than 2 mitoses/10 high-power fields); and (5)Pleomorphism should be considered malignant[39]. However, only evidence of local recurrence and distant metastasis represents unequivocal evidence of malignant behaviour[36].
Treatment:Due to the rarity of these neoplasms, no widespread guidelines are currently available. However, it is reasonable to use a conservative approach in cases of asymptomatic OGCTs measuring less than 1 cm, while ESD or surgical resection should be performed for symptomatic, larger tumours or when malignancy is suspected[37].
Oesophageal inflammatory myofibroblastic tumour
Background:Inflammatory myofibroblastic tumour (IMT) is defined as a distinctive lesion composed of myofibroblastic spindle cells accompanied by an inflammatory infiltrate of plasma cells, lymphocytes and eosinophils[40]. Oesophageal inflammatory myofibroblastic tumours (OIMTs) are exceedingly rare entities, with only a few cases reported in English-language medical literature[41,42].
Clinical features and diagnostic approach:OIMTs are usually symptomatic, and patients generally present with dysphagia, eventually accompanied by chest pain,globus sensation, postprandial emesis, regurgitation or cough; less commonly, weight loss and recurrent fever may also be present[40]. Even if it is not completely clear whether OIMTs are benign or malignant lesions, distant metastases and diseaserelated deaths have never been reported; hence, to date, it is possible to state that OIMTs have a benign behaviour[40]. The approach to OIMTs should be focused on a precise histopathologic diagnosis, the exclusion of multifocal disease and, finally, a treatment option that is as non-invasive as possible. Even if EUS and contrastenhanced CT scans may be helpful, the final diagnosis is usually made postoperatively after histological analysis of the resected mass.
Pathological features:The typical histological appearance of IMTs consists of spindled myofibroblasts and ganglion-like cells set in a fibromyxoid background associated with an inflammatory infiltrate composed of B lymphocytes, plasma cells and sometimes eosinophils (Figure 1E)[40]. On immunohistochemistry, IMTs are typically positive for SMA. More than 50% of paediatric cases are positive for ALK,indicating the presence ofALKgene rearrangement. Furthermore, CD117, CD34 and epithelial membrane antigen (EMA) are consistently negative[43].
Treatment:Surgical removal of OIMTs is considered the main treatment modality. In advanced disease, the therapeutic approach also involves systemic therapies such as chemotherapy, non-steroidal anti-inflammatory drugs, corticosteroid medications and targeted molecular therapy (effective in tumours with ALK rearrangement)[44-47].
RARE PRIMITIVE OESOPHAGEAL MALIGNANT TUMOURS OF EPITHELIAL ORIGIN
Oesophageal adenoid cystic carcinoma
Background:Oesophageal adenoid cystic carcinomas (OACCs) are rare, with slightly more than 100 cases reported in the medical literature[8,48-50]. OACCs are more common in Asians and generally affect men in their seventh decade of life[48,49]. OACCs typically occur in the middle third of the oesophagus[50].
Clinical features and diagnostic approach:Although superficial and asymptomatic OACCs have been discovered by routine EGDS, the most common symptom warranting further investigation is progressing dysphagia[50]. As is true for other oesophageal malignancies, EGDS with biopsy is necessary to confirm the diagnosis,and radiological evaluation with EUS, CT, MRI or PET permits clinical staging of the carcinoma[48-50].
Pathological features:Microscopically, OACCs consist of a mixture of epithelial and myoepithelial cells organized in cribriform, tubular or solid patterns. The cribriform pattern is characterized by tumour cells forming cystic lumina containing basophilic glycosaminoglycans (Alcian blue-positive) and hyalinized material (PASD-positive).In the cribriform and tubular patterns, the glands are lined by inner epithelial cells and outer myoepithelial cells, while in the solid pattern, no lumina are seen[8].Immunohistochemical staining methods are useful for diagnosis, demonstrating the two components of OACCs: Myoepithelial cells are positive for p63, S100, SMA and calponin, whereas epithelial cells are positive for cytokeratins, CEA and CD117[50,51].
Treatment:Complete surgical excision should be the first-line therapy, but this is seldom applicable: In the majority (76%) of cases, patients have distant metastases[48,50].Chemotherapy and radiotherapy are used both as adjuvant/neoadjuvant and palliative therapy, but their efficacy remains uncertain[50]. Despite the current treatments, the prognosis is poor, with a 1-year survival rate of 23%[48].
Oesophageal adenosquamous carcinoma
Background:Adenosquamous carcinoma is a malignant neoplasm composed of separate and coexisting elements of squamous cell carcinoma and adenocarcinoma[8].Oesophageal adenosquamous carcinomas (OASCs) are rare, with only a few dozen cases reported in the medical literature. OASCs are more common in male patients,with a median age of onset of 60 years[8,52], and the most common location is the middle third of the thoracic oesophagus[52]. The risk factors are similar to those for OSCCs[8].
Clinical features and diagnostic approach:The symptoms are similar to those of other oesophageal malignancies: Dysphagia, weight loss and retrosternal pain. The diagnostic approach is also analogous to that of the most common oesophageal carcinomas (i.e., EGDS with biopsy sampling and radiological exams)[8].
Pathological features:Microscopically, OASCs are composed of an admixture of both adenocarcinoma and squamous cell carcinoma. The proportion of the two components is generally considered insignificant to make the diagnosis of OASC.Nevertheless, the Japanese Classification of Oesophageal Cancer requires that each component represents at least 20% of the tumour; otherwise, the neoplasm is classified according to the predominant component alone[8]. The adenocarcinomatous component is typically tubular or glandular with mucin production, while the squamous areas can show any grade of differentiation[8,52]. Squamous and glandular components can be both intermixed or apparently separated, and the squamous elements are typically more superficially located than the glandular component[52].
Treatment:For eligible patients, the first-line treatment is surgical oesophagectomy with two-field lymphadenectomy, and patients who undergo adjuvant/neoadjuvant chemotherapy and radiotherapy have a relatively better survival than those who do not[52]. OASCs are thought to be more aggressive than classic OACs and OSCCs, with an overall five-year survival of 22.9%[52].
Oesophageal mucoepidermoid carcinoma
Background:Mucoepidermoid carcinoma is the most common malignant neoplasm of the salivary glands, while primary oesophageal mucoepidermoid carcinomas(OMECs) are uncommon, accounting for less than 1% of all cases of oesophageal carcinoma[53]. Patients are generally male (male to female ratio of 3:1) with a median age of 58 years and with a primary lesion located in the middle and lower third of the oesophagus[8,53].
Clinical features and diagnostic approach:Clinical presentation and diagnostic approach are comparable to those of other oesophageal malignancies. Biopsy is imperative to make the correct diagnosis[53].
Pathological features:The origin of OMECs is unclear: The most accepted hypothesis is that these neoplasms arise from a stem cell of the oesophageal glands or ductal cells with biphasic differentiation. On the other hand, some authors believe that progenitor cells are of squamous epithelial origin[8,53]. Microscopically, OMECs are composed of an intimate mixture of glands and nests of squamous cells and mucin-containing epithelial cells[8,53]. Immunostaining for p63 and CK7 can highlight squamous and mucin-producing cells, respectively[8].
Treatment:Surgical resection is the primary therapy in patients who can endure the operation[53]. The role of chemotherapy and radiotherapy is debated: Some studies suggest that neither chemotherapy nor radiotherapy is effective against OMECs,while others suggest that adjuvant chemoradiotherapy might prolong survival rates[53-55]. OMECs are more aggressive than OSCCs, and the prognosis is poor, with a five-year survival rate lower than 30%[53].
Oesophageal verrucous carcinoma
Background:Oesophageal verrucous carcinoma (OVC) is a rare variant of OSCC that typically arises in the lower oesophagus. OVCs usually affect the middle-aged population, with a male to female ratio of 2:1[56,57]. OVC risk factors are similar to those of classic OSCCs. The role of human papilloma virus (HPV) in the carcinogenesis of OVCs is controversial, but the most recent acquisitions suggest that HPV infection is probably not involved in OVC development and progression. On the other hand,TP53 gene mutation,EGFRoverexpression, and E-cadherin loss might fuel tumour proliferation and metastatic potential[58].
Clinical features and diagnostic approach:The most common symptoms in patients with OVCs are dysphagia and weight loss. Other reported symptoms include odynophagia, haematemesis and coughing[56,57]. EGDS is usually performed, showing OVCs’ typical endoscopic appearance: Wart-like lesions, even if polypoid features have been described[56]. EUS, CT and PET are useful tools for the clinical staging of patients. A correct preoperative diagnosis is often challenging and requires multiple deep biopsies together with a high grade of suspicion[56,57].
Pathological features:Microscopically, verrucous carcinomas have a heavily hyperparakeratinized epithelium or an irregular clefted surface with keratin plugging extending deeply into the clefts, usually associated with subepithelial inflammation.Cytologic atypia is minimal, and only tumour infiltration beyond the superficial mucosa can differentiate OVCs from a benign squamous lesion[57,59].
Treatment:In early-stage OVCs, ESD or surgical resection are first-line therapies, and in some cases, they can be curative[57,60]. In advanced cases, surgical treatment combined with radio/chemotherapy can be used, but the prognosis is usually poor[57].
Oesophageal carcinoma cuniculatum
Background:Carcinoma cuniculatum is a rare variant of squamous cell carcinoma that most commonly arises in the skin, but it may occur in other organs. For OSCCs,smoking is thought to be an important risk factor[61]. Oesophageal carcinoma cuniculatum (OCC) tumours are extremely rare, with just 17 cases reported in the medical literature[61]. Since pure forms of carcinoma cuniculatum tumours seem to have a better prognosis than OSCCs, it is important to recognize those entities in order to decide the most suitable treatment for the patient.
Clinical features and diagnostic approach:In patients with OCCs, dysphagia is the most common symptom, followed by regurgitation and diarrhoea[61]. CT scans and other radiological exams are useful to investigate patients preoperatively, but the correct diagnosis is usually made by postoperative histological analysis[61,62]. Biopsy examination may be useful; however, due to technical limitations of this procedure,the risk of misdiagnosis is high[61].
Pathological features:OCCs present a typical histological pattern characterized by hyperkeratosis, acanthosis, dyskeratosis, deep keratinization, keratin-filled cysts/deeply penetrating barrows, pseudo-koilocyte cells, intraepithelial neutrophil micro-abscess and mild cytologic atypia. The barrows are lined by very welldifferentiated squamous cells with absent to minimal cytologic atypia, and the surrounding stroma presents an inflammatory infiltrate rich in lymphocytes and plasma cells[61-63]. It is mandatory to exclude the presence of intermixed foci of conventional OSCC or OVC to rule out the diagnosis of so-called hybrid carcinoma,which has a worse prognosis than pure OCC[61].
Treatment:Due to the local aggressive behaviour of OCCs, radical surgery is necessary and is generally curative. Post-treatment prognosis seems to be favourable since just one patient of the 17 reported patients had a recurrence and died of cancerrelated death[61].
Oesophageal neuroendocrine tumours and carcinomas
Background:Neuroendocrine neoplasms (NENs) are a heterogeneous group of neoplasms originating from the diffuse neuroendocrine system. NENs are characterized by highly variable biological behaviour ranging from slow growing and low-grade malignant diseases to extremely aggressive diseases[64,65]. According to the 2019 WHO classification[8], the term “NEN” includes: (1) Well-differentiated neuroendocrine tumours (NETs); (2) Poorly differentiated neuroendocrine carcinoma(NEC); and (3) Mixed neuroendocrine-non-neuroendocrine neoplasm (MINEN), an umbrella category that includes mixed adenoneuroendocrine carcinoma. NENs of the oesophagus are very rare, accounting for less than 1% of all oesophageal malignancies, with an incidence that is higher in males (male to female ratio is 6:1)and a predominance in the sixth and seventh decade of life[66-69]. Approximately 90% of oesophageal NENs are NECs[70].
Clinical features and diagnostic approach:Patients typically present with dysphagia,weight loss, asthenia and sometimes with melena[67,68,70]. Metastatic NETs may also present with carcinoid syndrome[67]. Considering the clinical presentation, EGDS with biopsy sampling should be the first-line evaluation procedure: On endoscopy,oesophageal NETs appear as polypoid or nodular submucosal masses, while oesophageal NECs appear as large, infiltrative and ulcerated lesions[68,71,72]. The diagnostic assessment is completed by EUS, contrast-enhanced CT and MRI[73].
Pathological features:Microscopically, NETs consist of medium-sized cells with a low nuclear/cytoplasm ratio and a small ovoid nucleus with dispersed chromatin and small nucleoli. The growth pattern may be insular or cribriform, and a variable amount of well-vascularized stroma is present[8]. Immunohistochemical staining for common neuroendocrine markers (i.e., chromogranin A and synaptophysin)definitively confirms the diagnosis, while immunoreaction for Ki-67 is required for tumour grading[8]. NECs usually show solid, rosette-like and palisading patterns and can be subdivided into small-cell and large-cell NECs (Figure 1A). Small-cell NECs are characterized by small- to medium-sized cells with a high nuclear/cytoplasm ratio, scant basophilic cytoplasm and elongated nuclei with finely dispersed chromatin lacking well-formed nucleoli. Large-cell NECs are instead composed of medium- to large-sized cells with large ovoid nuclei containing large nucleoli. By definition, NECs are considered high-grade neoplasms[8]. Oesophageal MINENs usually consist of a NEC and either an OSCC or an OAC component; both neuroendocrine and non-neuroendocrine elements have to represent at least 30% of the tumour[74].
Treatment:The treatment of oesophageal NENs includes chemotherapy,radiotherapy, and surgery[64]. It is widely believed that the choice of therapeutic approach should mainly depend on clinical staging, distinguishing limited and extensive disease[64]. An oesophageal NEN is thought to be confined when it involves only the oesophagus and the surrounding tissues with or without local lymph node metastasis. In contrast, extensive disease affects distant organs and/or lymph nodes[64]. The combination of radical resection, radical lymph node dissection and chemotherapy may improve the prognosis of patients with limited lesions[75,76],whereas systemic chemotherapy is the only available treatment option for patients suffering from extensive disease[77]. In the case of small-cell NECs, because of the high expression of somatostatin receptors, somatostatin analogues such as 111Inoctreotide, 117Lu-octreotide, 90Y-octreotide, and 90Y-lanreotide may be effective in improving patient outcomes[78]. Finally, in recent years, a few clinical trials[79]have suggested the possible therapeutic value of molecular-targeted drugs inhibiting angiogenic mechanisms (e.g., bevacizumab) and various growth factor receptors expressed by oesophageal NENs (e.g., MAP kinase pathway inhibitors). Nevertheless,despite the aggressiveness of the aforementioned therapeutic approaches and the current advances in research, the prognosis of patients suffering from NECs remains dismal, with a median 5-year survival rate ranging from 0% to 27%, depending on the stage and type of treatment[77].
Extramammary Paget’s disease of the oesophagus
Background:Paget’s disease is thought to be a peculiar type of epidermotropic adenocarcinoma of mammary and extramammary tissues[80]. Extramammary Paget’s disease is most commonly seen in the vulva and anus, while an oesophageal location is exceedingly rare, with less than ten cases described[81,82]. Oesophageal Paget’s disease (OPD) always arises in the setting of Barrett’s oesophagus[81,82].
Clinical features and diagnostic approach:Typically, patients complain of progressive dysphagia, eventually accompanied by chest discomfort similar to reflux oesophagitis[81,82]. EGDS is a first-line diagnostic tool. Since the gross appearance of OPD may mimic a common inflammatory lesion, Lugol chromoendoscopy should be applied to help with a correct diagnostic framework[83]. Biopsy should always be performed since the diagnosis of OPD is ultimately histopathological.
Pathological features:The microscopic features of OPD include the presence of intraepithelial nests and isolated cells with large round-shaped cytoplasm spreading within the entire squamous epithelial layer. These cells usually invade the mucosal and submucosal ducts multifocally, indicating adenocarcinomatous differentiation[82].One of the main differential diagnoses is primitive oesophageal melanoma: Positive immunohistochemistry staining for mucins, cytokeratin 18 and E-cadherin, together with negative staining for S100, will help distinguish OPD from oesophageal melanoma[82].
Treatment:Considering the strong association between OPD and the presence of an aggressive and poorly differentiated carcinoma, patient management should be the same as that adopted when a diagnosis of high-grade carcinoma is made.
RARE PRIMITIVE OESOPHAGEAL MALIGNANT TUMOURS OF NON-EPITHELIAL ORIGIN
Primitive oesophageal melanoma
Background:Oesophageal melanomas (OMs) have an estimated incidence of 0.03 cases per million in the United States, accounting for 3% of all melanomas and for 0.2% of all collective oesophageal malignancies[84,85]. Both the risk factors and the pathogenesis of OMs are still debated. Although melanocytes may encase the oesophageal mucosa as a consequence of aberrant early migration of melanoblasts from the neural crest[86], individual and environmental drivers of malignant degeneration of these cells are still unclear[85,86]. Moreover, the multiphasic molecular pathway underlying the development of OMs is still uncharacterised.BRAFp.V600Emutation, commonly seen in approximately 50% of cutaneous melanomas[87], has never been described in OMs. Furthermore, mutations in theSFB1,GNAQorGNA11genes, often observed in uveal melanomas, were not found in OMs with an oesophageal location[85]. OMs have been described to harbour activating mutations of IGF1R (82% of cases), KIT overexpression (50% of cases) and constitutive activation of MAPK[85](Table 1).
Clinical features and diagnostic approach:Patients with OMs are more often men in their 5th-6thdecade presenting with dysphagia, malaise, anaemia and retrosternal pain[84]. The subsequent endoscopic exploration of the upper digestive tract results in the identification of neoplastic strictures (generally appearing as large brown masses with tiny brownish satellite patches) usually situated in the middle third or lower third of the oesophagus, while the involvement of the cervical segment is rarer[85]. To state the true origin of the lesion, careful clinical, endoscopic, radiologic and pathological investigations are required.
Pathological features:The histological pattern of OMs consists of nests of epithelioid or spindle-shaped cells with prominent nucleoli; isolated malignant melanocytes can be seen, and necrosis may be present (Figure 1B)[85]. Positive immunostaining for melan A and HMB45 confirms the melanocytic nature of the cells with high sensitivity and specificity[85]. Moreover, it is useful to assess resection margins since in most cases, the tumour is more extensive microscopically than is seen on gross examination of the resected specimen. By contrast, the differential diagnosis between primary or metastatic OMs is extremely challenging: The presence of melanocytes at the epithelial-stromal junction and/or the recognition of adjacent melanoma in situ can suggest a primary lesion, but an extensive clinical examination is mandatory for the final diagnosis[88].
Treatment:The therapeutic approach to OMs depends on the tumour stage at presentation and the intrinsic biological behaviour of the malignancies[85]. Regardless,OM prognosis is very poor, with a 5-year overall survival rate of approximately 5%[88].Adequate resectional surgery seems to be the only chance of long-term survival, and it should always be combined with different adjuvant therapies, such as chemotherapy, immune stimulation, radiotherapy and photo-radiation[85].
Oesophageal gastrointestinal stromal tumour
Background:Gastrointestinal stromal tumours (GISTs) are the most common mesenchymal tumours of the gastrointestinal tract[89]. They are thought to result from the neoplastic degeneration of a precursor cell with differentiation towards the Cajal cell phenotype[90]. Oesophageal GISTs (OGISTs) are extremely uncommon,representing less than 1% of all GISTs[91]. One of the characteristic “molecular signs” of GISTs is the activating mutations ofKIT, which are now considered the causal molecular events for the development of these neoplasms[8,92]: This is of great therapeutic interest since targeted tyrosine-kinase inhibitors are currently used in the treatment of selected patients suffering from GISTs.

Table 1 Main histological patterns and key immunophenotypic/molecular markers in oesophageal sarcomas
Clinical features and diagnostic approach:GISTs usually affect middle-aged males.The most common symptom at onset is represented by dysphagia (to both liquids and solids). Other possible symptoms often include epigastric pain and bleeding, while weight loss is less frequent[93]. The biological behaviour of GISTs is variable: A properly benign attitude can be observed in 70%-90% of cases, while approximately 10% to 30% of these neoplasms progress to malignancy[94]. Hence, the major issue in the management of GISTs is both the formulation of the diagnosis and the estimation of the malignant potential of the lesion. To attain these goals, a comprehensive approach to the patient, including clinical, endoscopic, radiologic and histopathological investigations, is warranted (Figure 2C).
Pathological features:Microscopically, GISTs appear as well-defined and pseudoencapsulated lesions adherent to the muscularis propria (Figure 1D). The growth pattern can be both intraluminal and eccentric (i.e., mediastinal outgrowth)and foci of necrosis may be present. The cellular phenotype ranges from spindleshaped to epithelioid[93,94]. Approximately 98% of GISTs are immunoreactive for CD117 and DOG1, which represents the diagnostic hallmark for these lesions. Other immunohistochemical markers that can be expressed in GISTs are CD34, SMA,desmin and S100[93,94]. Mitotic count per 5 mm2area, tumour size and anatomic site are required parameters to assess the patient’s prognosis[95-97]. Furthermore, intrabdominal capsule rupture is associated with an extremely high risk of local recurrence that negatively impacts prognosis[8].
Treatment:The matters related to the therapeutic approach of OGISTs are complex and fall outside the scope of this review. Briefly, the management of each individual patient should be discussed and agreed upon by a multidisciplinary team, taking into account the size, location and biological features of the neoplasm together with the patient’s surgical risk. In principle, complete surgical resection is the best treatment for localized GISTs that can be resected without excessive risk or subsequent functional deficit. Surgical options include either enucleation or oesophagectomy, and both show further positive outcomes when associated with neoadjuvant and/or adjuvant molecularly targeted therapy[89,98].
Oesophageal liposarcoma
Background:Liposarcomas are rare mesenchymal malignancies. Oesophageal liposarcomas (OLSs) are extremely rare, with approximately 40 cases reported in the medical literature[99]. OLSs seem to occur preferably in middle-aged to elderly male patients[99]. OLSs can be classified into three subtypes: (1) Well-differentiated and dedifferentiated liposarcoma (WDLS/DDLS); (2) Myxoid liposarcoma (MLS); and (3)Pleomorphic liposarcoma (PLS)[100]. The pathogenesis of liposarcomas is still unclear,but some molecular alterations specific to each subtype have been described and are thought to be involved in the tumorigenesis process. In WDLSs/DDLSs, there is evidence of the oncogenic role ofMDM2, CDK4, HMGA2andTSPAN31[101]. In MLSs,reciprocal translocation occurs between chromosomes 12 and 16, leading to fusion of theDDIT3andFUSgenes and activating some downstream targets, such asPPARgamma2andC/EBPalpha, which promote cell cycle proliferation[101]. PLS is the most complex and least understood subtype of three: Mutations in various tumour suppressor pathways components, such asTP53,NF1andRB1, account for the aggressive nature of this tumour[102].
Clinical features and diagnostic approach:Since OLSs occur more frequently in the submucosal layer of the upper oesophagus, symptoms are related to this localization[102]. Patients usually present with dysphagia, cough, vomiting, foreign body sensation and weight loss[102]. Long-standing OLSs can have serious complications, such as anaemia, weight loss, regurgitation-associated aspiration with asphyxiation and respiratory failure[103]. Patients should be studied with EGDS and many radiological exams, particularly with CT and MRI. The definitive diagnosis,however, is still histopathological[99].
Pathological features:The histological classification of liposarcomas is heterogeneous. The most typical variant of WDLS is composed of a mature fat component of variable size associated with fibrous septa, including atypical spindle cells with enlarged and hyperchromatic nuclei (the so-called adipocytic variant)(Figure 1G). Mitotic figures are rare. The amount of lipoblasts is variable, most often not significant and not required for the diagnosis of WDLS[104]. DDLSs have a wide morphological spectrum. The tumours appear like undifferentiated pleomorphic or spindle-cell sarcomas. DDLSs typically show moderate to high cellularity with mild to severe atypia. The pattern of growth can be patternless, in fascicles or storiform in architecture. The stroma can appear collagenous, myxocollagenous or myxoid. Welldifferentiated and dedifferentiated components are both generally present and can have abrupt or gradual transitions[104]. Almost all WDLSs and DDLSs are characterized molecularly by the presence ofMDM2andCDK4gene amplification[104].
MLSs are typically composed of bland fusiform to ovoid cells in a myxoid stroma with a prominent plexiform capillary network (i.e. chicken-wire vasculature) and scattered monovacuolated lipoblasts[100]. MLSs are generally S100 positive andMDM2negative; FISH analysis showsDDIT3rearrangement, which represents a very useful finding for the diagnosis[100]. PLSs are composed of pleomorphic cells, with highly atypical enlarged and bizarre nuclei and often prominent nucleoli, associated with usually numerous multivacuolated lipoblasts, which represent a diagnostic clue[105].Less frequently, the tumour can be composed of round, spindle or epithelioid cells.Necrosis and mitoses are common. On immunohistochemistry, CD34 staining can be strongly positive, but this marker is not specific[105].
Treatment:Surgical excision is the mainstay of treatment[106]. Depending on the size of the tumour, surgery can be either minimally invasive ESD[107]or aggressive partial or total oesophagectomy. The role of adjuvant chemotherapy and radiation therapy is controversial, and long-term follow-up is recommended due to the high rate of recurrence[99].
Oesophageal malignant peripheral nerve sheath tumour
Background:The classification malignant peripheral nerve sheath tumour (MPNST)was introduced in 2002 by the World Health Organization to encompass all malignancies arising from the peripheral nervous system or that show nerve sheath differentiation[108]and fulfil at least one of the following criteria: (1) Peripheral nerve origin; (2) Pre-existing benign nerve sheath tumour origin; (3) Histological demonstration of Schwann cell differentiation; and (4) Being a malignant spindle cell tumour in a patient with neurofibromatosis type 1, until proven otherwise[109].MPNSTs are very uncommon diseases, accounting for 3%-10% of all soft tissue sarcomas, and in 50% of cases, they develop in the setting of neurofibromatosis type 1[108]. In this landscape, oesophageal MPNSTs (OMPNSTs) are rarer still, and to date,only fourteen cases have been reported in the medical literature[110]. OMPNSTs should be considered unique entities, since their biologic behaviour and clinical prognostic features are quite different from those of MPNSTs developing elsewhere throughout the body. Although MPNSTs are generally considered aggressive malignancies with high recurrence rates after surgical treatment and poor prognosis, previous reports of cases with oesophageal localization have recorded that early-stage disease, often found at first presentation, shows satisfactory outcomes[111]. OMPNSTs are also characterized by a specific epidemiologic pattern, being relatively more common in females than in males (male to female ratio 4:10)[112]and in Asians than in people from other parts of the world[111]. Finally, it is of great interest that OMPNSTs (as well as other gastrointestinal MPNSTs) seem to be sporadic, since the association with NF-1 has never been observed[113].
Clinical features and diagnostic approach:Patients with OMPNSTs usually present with progressive dysphagia, epigastric discomfort, heartburn, fatigue and weight loss.Physical examination is generally unremarkable, with the exception of occasional signs of anaemia[110]. The first-line EGDS often results in the identification of a large(median size of 6 cm in diameter) solid and greyish submucosal lesion in the distal oesophagus with an ulcerated surface[110]. Preoperative assessment should be completed by means of radiologic exams to obtain information about the depth of neoplastic infiltration into the oesophageal wall, the involvement of regional lymph nodes and the presence of distant metastases[110]. The final diagnosis is histopathological.
Pathological features:Microscopically, MPNSTs appear as monomorphic malignant mesenchymal neoplasms composed of spindle cells arranged in intersecting bundles[110]. Variations in cellularity and cellular condensation around blood vessels are the most distinctive morphological features. The cells have eosinophilic and basophilic cytoplasm. A high number of mitoses and extensive necrotic foci are typically present. All these features help in the differential diagnosis from other oesophageal sarcomas, such as leiomyosarcomas and high-grade GISTs and occasionally metastatic desmoplastic melanoma[109,110,112,114]. On immunohistochemistry,the tumour cells are negative or focally positive for S100[110]. In some cases, divergent rhabdomyoblastic differentiation can be appreciated both morphologically and immunohistochemically with the expression of desmin and myf4. In this scenario, the tumour is also called malignant triton tumour[12].
Treatment:Radical surgical resection is the treatment of choice, and surgical options include both tumour enucleation and oesophagectomy, eventually with total gastrectomy and regional lymphadenectomy[110]. In unresectable or metastatic cases,palliative treatments include palliative surgery and adjuvant therapies such as radiotherapy and chemotherapy[110].
Oesophageal perivascular cell tumour
Background:PEComas are a family of rare mesenchymal neoplasms that can develop in many locations throughout the body, such as the lung (e.g., pulmonary sugar tumour and lymphangioleiomyomatosis), kidney (e.g., angiomyolipoma), falciform ligament (e.g., clear cell myomelanocytic tumour) and soft tissue or bones[115,116]. All these tumours are linked by the proliferation of perivascular epithelioid cells and a common genetic background (i.e., alterations in the tuberous sclerosis gene complex)[117,118]. Oesophageal PEComas (OPEComas) are extremely rare, with just one case reported in the medical literature[115]. The biological behaviour of these neoplasms can be very variable, and PEComas are classified as benign, of uncertain malignant potential, or malignant on the basis of the following features: (1) Tumour size > 5 cm;(2) Infiltration; (3) High nuclear grade; (4) Increased cellularity; (5) High mitotic activity (≥ 2 mitotic figures/50 HPFs); (6) Tumour necrosis and (7) Vascular invasion.Benign PEComas have none of these features, while PEComas with uncertain malignant potential exhibit one; when 2 or more worrisome features are noted, the diagnosis of malignant PEComa is made[119].
Clinical features and diagnostic approach:Because of the exceptional rarity of the oesophageal location of a PEComa, this diagnosis can be made only after resection of the entire tumour mass. Indeed, the symptoms (i.e., dysphagia), macroscopic aspects and radiologic features of a PEComa arising in this site are indistinguishable from those of other submucosal lesions of the oesophagus.
Pathological features:Microscopically, the only reported oesophageal PEComa appeared as a diffuse proliferation of polygonal/spindle cells with a nonspecific growth pattern; this is in contrast with the typical radial arrangement of neoplastic cells around the vascular lumen seen in other PEComas[115]. The cytoplasm is variably abundant and clear to granular, and cells have round to oval nuclei. On immunohistochemistry, PEComas are positive for HMB45, S100, vimentin and MIFT and negative for common markers of epithelial, neuroendocrine or mesenchymal origin[115].
Treatment:Surgical excision is the first-line treatment. The only patient diagnosed with an OPEComa died three months after the initial diagnosis after a devastating course of neoplastic disease and progressive physical deterioration[115].
Primitive oesophageal mucosa-associated lymphoid tissue lymphoma
Background:The gastrointestinal tract is the most common extranodal site of lymphoma, the majority of which is classified as mucosa-associated lymphoid tissue(MALT) lymphoma, a low-grade malignant non-Hodgkin B cell lymphoma[120]. The stomach is the most common location of MALT lymphoma, while primary oesophageal MALT lymphoma is extremely rare; there are only 17 reported cases of this disease, most of which have been observed in Japan[121,122]. The pathogenesis of these rare malignancies is widely unknown[123]: While it is established that 80% to 90%of gastric MALT lymphomas are associated withHelicobacter pyloriinfection[124], there is no evidence of this association in oesophageal MALT lymphomas. Nevertheless, it is reasonable to acknowledge an aetiologic role of chronic inflammation in oesophageal MALT lymphomas, since structured lymphatic tissue or follicles are lacking in normal oesophageal mucosa, but they may appear in the setting of chronic inflammation[123].
Clinical features and diagnostic approach:The most common presenting symptom is dysphagia. Of note, MALT lymphomas involving the proximal oesophagus are frequently complicated by haemorrhage, obstruction and perforation with fistula formation, which often results in rapid death secondary to aspiration pneumonia[120,121]. The diagnosis of oesophageal lymphoma requires the integration of data from clinical, endoscopic, endosonographic and radiologic studies. Nevertheless,the final diagnosis depends upon detailed histological, immunohistochemical and gene rearrangement analyses[121]. Endoscopically, oesophageal MALT lymphoma appears as a submucosal lesion usually located in the middle or lower third of the organ[121,125].
Pathological features:Morphological characterization reveals neoplastic lymphocytes with moderate atypia, clear cytoplasm and irregular nuclear membrane that proliferate with a diffuse pattern in the submucosal layer. Neoplastic lymphocytes can be classified as centrocyte-like cells, monocyte-like cells and small lymphocyte-like cells. These three types can exist alone or can be admixed in different proportions[121].Interestingly, so-called lymphoepithelial lesions, which are a hallmark of gastric MALT lymphoma, have rarely been observed in oesophageal locations[121]. The assessment of clonal rearrangement of immunoglobulin light and heavy chains definitively confirms the diagnosis of lymphoma, while several recurrent chromosomal abnormalities, such as t (11; 18) (q21; q21), t (1; 14) (p22; q32), t (14; 18)(q32; q21), t (3; 14) (p14.1; q32), and trisomies 3 and 18, are prognostic and predictive markers[124,126-131].
Treatment:The best therapeutic approach to oesophageal MALT lymphoma is still an open issue. In general, the management depends on histology, stage at presentation,clinical characteristics before the initial treatment and coexistent complications[125].Chemotherapy, radiotherapy, endoscopic resection and convectional surgery are used with similar success[125]; in fact, irrespective of the treatment modality, the prognosis is good since MALT lymphoma is a low-grade malignant tumour[121].
Oesophageal solitary fibrous tumour
Background:Solitary fibrous tumours are mesenchymal neoplasms that occur in the visceral pleura in 85%-90% of cases. The tumours typically affect middle-aged adults without sex predilection[132]. Solitary fibrous tumour of the oesophagus is extremely rare, with fewer than ten cases reported in the medical literature[132-136]. This neoplasm usually has indolent behaviour, but 10%-15% of thoracic solitary fibrous tumours show more aggressive behaviour; one case of oesophageal solitary fibrous tumour(OSFT) with aggressive behaviour has been reported[133].
Clinical features and diagnostic approach:Patients with OSFT usually present with dysphagia, chest pain and/or increasing dysphonia[132-134,136]. EGDS, EUS and CT are useful for the correct clinical assessment, but the diagnosis is made after histological examination of a biopsy or surgical resection specimen[132,133,136].
Pathological features:Microscopically, solitary fibrous tumours are submucosal lesions composed of a patternless proliferation of spindle cells within a collagenous matrix with cracking artefacts and staghorn vessels. Features of malignancy are high cellularity, cellular pleomorphism, necrosis and more than 4 mitoses per ten highpower fields. STAT6-positive nuclear immunoreaction confirms the diagnosis. OSFT is strongly positive for CD34 and BCL-2, but the more specific and sensitive marker for the diagnosis of OSFT isSTAT6, which is expressed in almost all cases[12,133]. The expression ofSTAT6indicates the presence of a specific recurrent translocation involving theSTAT6andNAB2genes. The neoplasm is negative for S100.
Treatment:Endoscopic or surgical resection is considered curative for indolent OSFT[132,134,136]. The only malignant OSFT was treated with surgical enucleation, and adjuvant radiotherapy was performed; after 32 months, the patient was alive and without evidence of tumour recurrence[133].
Oesophageal leiomyosarcoma
Background:Leiomyosarcoma is the most common human sarcoma, and oesophageal location is rare, with oesophageal leiomyosarcoma (OLMS) accounting for less than 1% of oesophageal malignancies; nonetheless, leiomyosarcoma is the most common primitive oesophageal sarcoma[137-139].
Clinical features and diagnostic approach:Dysphagia is the most common symptom in patients with OLMS, but other symptoms are retrosternal backpain, weight loss and/or emesis[138]. OLMS is reported to arise from the muscular layer of the oesophagus. EGDS, EUS, CT and PET/CT are useful, but the final diagnosis is usually made after histological examination of an oesophageal biopsy[138].
Pathological features:Microscopically, leiomyosarcomas can present different morphologies. There are three main histological variants of leiomyosarcoma: A conventional/spindle cell type, a myxoid variant and an epithelioid variant. The conventional type is characterized by spindle cell proliferation with at least two of the following features: Marked cellular atypia, more than 10 mitoses per 10 high-power fields and the presence of necrosis. The myxoid variant is described as a hypocellular tumour with abundant myxoid stroma characterized by any grade of nuclear atypia,tumour cell necrosis or more than 1 mitosis per 10 high-power fields. The epithelioid variant is described to feature proliferation of round or polygonal cells with eosinophilic or clear cytoplasm arranged in nests, sheets or cords with moderate to severe cytologic atypia, tumour cell necrosis or more than 3 mitoses per 10 highpower fields[12]. On immunohistochemistry, leiomyosarcomas are positive for SMA(95%), muscle-specific actin (91%), calponin (88%), desmin (73%), caldesmon (66%)and myosin (64%) and negative for CD34, CD117, DOG1 and S100[137,140].
Treatment:ESD and radical surgical resection, eventually followed by adjuvant radiotherapy, are the first-line therapeutic approaches for OLMS[138-140]. OLMSs have a better prognosis than other oesophageal malignancies: In the Chinese medical literature, the 3-, 5- and 10-year survival rates are reported to be 80.0%, 58.3% and 31.1%, respectively[138].
Oesophageal synovial sarcoma
Background:Synovial sarcoma is a malignant mesenchymal tumour typically involving the soft tissue of the extremities. The digestive tract is an extremely rare location, and fewer than a dozen oesophageal synovial sarcomas (OSSs) are reported in the medical literature[141].
Clinical features and diagnostic approach:Dysphagia and weight loss are typical,although nonspecific, symptoms in patients with OSS[141,142]. EGDS, CT and positron emission tomography-computed tomography (PET-CT) are useful for the correct assessment of the patient, but the diagnosis is made postoperatively with histopathological examination[141].
Pathological features:Synovial sarcoma can be biphasic or monophasic. In biphasic synovial sarcomas, spindle cell and epithelial components are present in varying proportions. Epithelial components can be organized in nests or cords or with glandular, alveolar or papillary architecture, while the spindle cell component is arranged in dense cellular sheets or vaguely formed fascicles[12]. On immunohistochemistry, synovial sarcomas are positive for EMA, cytokeratins, CD99 and TLE-1.S100 is positive in 40% of cases, while positivity for CD34 is extremely rare[12]. The translocation t (X; 18) is characteristic of this synovial sarcoma, since it is found solely in this tumour[12].
Treatment:Surgical excision is the first-line therapy, eventually followed by chemotherapy[141-143]. Due to the rarity of this disease, solid data about the prognosis of OSS are not available in the medical literature.
Oesophageal Kaposi sarcoma
Background:Kaposi sarcoma is an uncommon, low-grade vascular neoplasm associated with HHV8 infection. There are four forms of Kaposi sarcoma: (1) The classic, usually nonprogressive variant, which is typically seen in elderly men from Eastern Europe and characterized by multiple purple nodules of the lower limbs; (2)The lymphadenopathy-associated form, which is very aggressive and endemic in Africa; (3) The post-transplant/immunosuppression-associated form; and (4) The acquired immune deficiency syndrome (AIDS)-associated form[144]. In 40% of cases,patients with Kaposi sarcoma have visceral or lymph node involvement, and the gastrointestinal tract is the most common location. The small intestine is most frequently affected, followed by the stomach, oesophagus and colon[145].
Clinical features and diagnostic approach:Oesophageal Kaposi sarcoma (OKS), like other Kaposi sarcomas of the gastrointestinal (GI) tract, is usually asymptomatic, but patients with upper gastrointestinal bleeding and pain have been reported[144,145].Histopathological examination of EGDS-obtained biopsies can be diagnostic, and radiological exams are useful for correct clinical staging[144,145]. The clinical history of the patient can be helpful for diagnosis.
Pathological features:Microscopically, Kaposi sarcoma is characterized by spindle cell proliferation forming vessels of irregular morphology or slits associated with extravasation of erythrocytes, haemosiderophages and a significant lymphoplasmacytic infiltrate[146]. On immunohistochemistry, Kaposi sarcoma expresses ERG,CD31, CD34, and HHV-8, with HHV-8 being 100% specific for Kaposi sarcoma[144,146].
Treatment:Visceral involvement of Kaposi sarcoma generally has a poor prognosis,and treatment is usually palliative. Therapeutic options include antiretroviral medications, radiation therapy, modification in the immunosuppressive regimen and chemotherapy with liposomal anthracyclines[144,146].
Oesophageal angiosarcoma
Background:Angiosarcomas are rare malignant tumours that usually arise in the skin, breast, liver, spleen and bones. Oesophageal angiosarcoma (OA) is extremely rare, with fewer than ten cases reported in the medical literature[147,148].
Clinical features and diagnostic approach:Patients affected with OA usually report severe and progressive dysphagia. EGDS can show ulcerative lesions, and biopsies are necessary for the diagnosis[147].
Pathological features:Microscopically, OAs are composed of capillary-like structures and sinusoidal spaces lined with atypical endothelial cells sometimes with epithelioid morphology. Immunohistochemically, OAs are positive for CD31 CD34, FLI1 and ERG. Interestingly, angiosarcomas express cytokeratins and synaptophysin, which in case of epithelioid morphology can represent an important diagnostic pitfall[12,147].
Treatment:Oesophagectomy is the mainstay treatment. OAs seem to have a more favourable prognosis than other non-oesophageal angiosarcomas[147].
Oesophageal Ewing sarcoma
Background:Ewing family tumours include Ewing sarcomas and peripheral primitive neuroectodermal tumours. Neoplasms with primitive neuroectodermal differentiation generally arise in the bones, chest wall, paravertebral region,retroperitoneum and lower extremities of children and young adults. Primitive Oesophageal Ewing sarcoma (OES) is extremely rare, with only a few cases reported in the medical literature[149,150]. More than 80% of these tumours are characterized by t(11; 22) (q24; q12), which results in theEWS-FLI-1fusion protein[12].
Clinical features and diagnostic approach:Dysphagia and progressive dysphonia are reported symptoms in patients with OES[149,150]. EGDS with bioptic sampling is necessary for diagnosis, while CT and PET are useful for clinical staging[149].
Pathological features:Microscopically, this neoplasm is composed of cells with small,uniform, mildly atypical nuclei with occasional nuclear grooves, granular chromatin and little cytoplasm arranged in trabecular and nested growth patterns with perivascular rosettes and pseudopapillary architecture. On immunohistochemistry,these tumours are positive for CD99, FLI-1, beta catenin, cyclin D1, p53 and NSE but negative for melan A and HMB45. FISH analysis for the t (11; 22) translocation is important to support the diagnosis[149].
Treatment:Due to the rarity of this condition, it is unclear which therapeutic approach should be carried out. Surgical resection should be performed when possible, eventually accompanied by chemotherapy[149,150].
FURTHER RARE OESOPHAGEAL PRIMITIVE MALIGNANT NEOPLASMS
To have a full picture that is as precise as possible of all the potential malignant neoplasms occurring within the oesophagus, it is necessary to report on other extremely rare histotypes. Primary oesophageal carcinosarcoma[151],rhabdomyosarcoma[152], chondrosarcoma[153], epithelioid sarcoma[154], diffuse large B cell lymphoma[155]and Hodgkin lymphoma[156]have been described. Unfortunately, for these tumours, no robust data about clinical and/or therapeutic approaches are available.
SECONDARY OESOPHAGEAL INVOLVEMENT OF MALIGNANCIES: HAEMATOGENOUS METASTASIS TO THE OESOPHAGUS
Blood-borne metastasis to the GI tract is uncommon, with the stomach and small bowel being the segments most frequently involved[157]. An oesophageal location of blood-borne metastases is even rarer: In a large series of autopsies performed in patients who died from advanced cancers collected by Mizobuchiet al[158], metastasis to the oesophagus was found only in 112 of 1835 cases (6.1%). Due to the rarity of this condition and the lack of large multi-centre case series, there is no agreement among authors on the epidemiologic impact of each type of secondary oesophageal malignancy. Malignant melanoma[157]and lung cancer[158]seem to be the most common tumours that can metastasize to the oesophagus.
Secondary oesophageal involvement occurs virtually always as a part of multipleorgan metastatic disease, thus resulting in poor prognosis[159]. Nevertheless, if the oesophagus is the only organ involved in some stage IV cancers (e.g., breast cancer),the oesophageal location does not seem to affect the prognosis of patients, even if the associated symptoms might significantly decrease their quality of life[159]. Hence,properly and rapidly diagnosing a secondary oesophageal neoplasm is imperative,since surgical resection may provide excellent palliative therapy and long survival[160,161]. Oesophageal metastasis should be considered in all patients complaining of dysphagia and diagnosed with any other malignant neoplasm.However, secondary oesophageal malignancies are usually clinically silent and are found incidentally during the post-mortem exam[158,160]. The diagnosis of oesophageal metastasis is difficult and requires strong clinical suspicion together with comprehensive knowledge of clinical, radiologic, endoscopic and pathological aspects. EGDS, barium thoracic radiography and explorative mediastinoscopy and surgery are generally required for the final diagnosis[158,162,163]. Of note is that when an oesophageal secondary lesion is recognized and the primary tumour is occult, a further work-up of the lung is always necessary[158].
On histopathological examination, all oesophageal metastases appear as infiltration of the submucosa and deeper layers of the organ wall by cancer cells, while the squamous epithelium is intact[158]. This is important for two reasons: (1) Every oesophageal stricture covered with normal mucosa in an oncological patient should be investigated as a metastatic tumour; and (2) The initial biopsy often fails to provide a sufficient amount of sample, since the mucosa above the lesion is typically intact[158].After the initial morphological exams, immunohistochemical staining usually provides the correct final diagnosis.
CONCLUSION
The aim of this review was to provide an overview of the uncommon neoplastic lesions of the oesophagus. Despite the exceeding rarity of these tumours, awareness of their existence and clinical presentation and adequate diagnostic-therapeutic followup are important. In fact, patient prognosis is often more subject to the prompt diagnosis and appropriateness of the clinical approach rather than the natural history of the disease itself. Nevertheless, some rare primitive oesophageal tumours, such as melanomas, retain a dismal prognosis irrespective of all therapeutic attempts, similar to the more common oesophageal carcinomas.
Major diagnostic AIDS in the approach to rare oesophageal neoplasms include contrast-enhanced CT, MRI, PET-CT and endoscopic techniques with biopsy sampling. Gaining a correct final diagnostic assessment, however, remains challenging largely due to the frequent submucosal location of the lesions, which is not accessible with common biopsy techniques. Hence, the final diagnosis is often established after the examination of the entire surgically removed lesion.
Regarding therapeutic approaches of almost all rare oesophageal neoplasms, there is a generalised lack of agreement between authors. Regardless of the consensus about the possibility of adopting a conservative/wait-and-see attitude towards small,benign and asymptomatic tumours, the best approach for symptomatic and/or malignant diseases is still debated. In the available medical literature, surgical,endoscopic, radiological and medical interventions have been proposed, but the evaluation of outcomes is almost always meaningless on account of the small number of patients treated (often less than 10). However, it is possible to state that the key factors considered when choosing a therapeutic approach are primitive tumour dimensions and presence of metastatic spread to distant organs or loco-regional lymph nodes. In cases of malignancies confined to the oesophagus, the therapeutic attitude (surgicalvsendoscopic) should be chosen with the aim of guaranteeing R0 resection, which correlates with lower recurrence risk.
Ultimately, primitive oesophageal neoplasms of uncommon histology should always be taken into account in patients presenting with dysphagia (to liquids and solids) and first EGDS non-diagnostic for cancer or common causes of benign stenosis.The correct diagnostic and therapeutic management of these diseases should be delegated to highly specialized centres and requires a multidisciplinary effort involving clinicians, endoscopists, radiologists, pathologists, surgeons and oncologists. Moreover, systematic collections of large case series are required to better evaluate outcomes and find promising therapeutic interventions derived from solid,evidence-based data.
 World Journal of Gastroenterology2020年27期
World Journal of Gastroenterology2020年27期
- World Journal of Gastroenterology的其它文章
- Details determining the success in establishing a mouse orthotopic liver transplantation model
- Modified percutaneous transhepatic papillary balloon dilation for patients with refractory hepatolithiasis
- Serum ceruloplasmin can predict liver fibrosis in hepatitis B virusinfected patients
- Acceptance on colorectal cancer screening upper age limit in South Korea
- Transarterial chemoembolization with hepatic arterial infusion chemotherapy plus S-1 for hepatocellular carcinoma
- Intestinal NK/T cell lymphoma: A case report