
食品中Escherichia coli O157:H7微滴数字PCR绝对定量检测方法的建立
2020-08-26 03:50魏咏新徐蕾蕊魏海燕张西萌
食品科学 2020年16期
魏咏新,马 丹,李 丹,徐蕾蕊,魏海燕,张西萌,刘 莉,曾 静*
(北京海关技 术中心,北京 100026)
大肠埃希氏菌(Escherichia coli)O157:H7是一种重要的人兽共患传染病原菌[1-2],是肠道出血性E. coli中对人致病性最强的血清型之一,以食物为主要的传播途径,牛、羊、猪 和鸡等家畜家禽为主要宿主[3],其产生的细菌毒素,可引起出血性结肠炎,可发展成溶血性尿毒综合症和血小板减少性紫癜,严重者可致死亡[4]。世界上多个国家都曾有过不同规模的E. coli致病事件爆发和流行,1982年美国首次报道了由E. coliO157:H7引发的出血性肠炎,紧随其后加拿大、日本、英国等国也接连报道了E. coliO157:H7的爆发[5-7]。我国江苏、安徽省也曾经在1999年爆发E. coliO157:H7感染性腹泻,超过2万 例患者,死亡177 例,流行时间达7 个月之久[8]。
因为致病菌长期与人类共存,国内关于食品中致病菌存在和计数要采用危险性评估的方法代替零容许值的问题正引起日益激烈的争论,在这种情况下致病菌定量检测方法尤为重要[9]。致病菌定量快速检测对于执行食品微生物限量标准、对食品中的致病微生物进行风险评估更具有实际的意义[10]。GB 4789.36—2016《食品微生物学检验 大肠埃希氏菌O157:H7/NM检验》[11]采用传统分离培养法(或免疫磁珠捕获法)、生化确认与血清学实验结合的检测方法,检测周期长,操作复杂且只能实现定性检测。目前我国还没有制定E. coliO157:H7定量检测标准,因此本研究旨在建立食品中E. coliO157:H7的绝对定量检测方法,对E. coliO157:H7的污染水平提供数据依据,为食品安全检测、开展风险评估等工作提供有效的思路和借鉴。
现有常用的致病菌定量检测方法包括平板法、最大可能数(maximum probable number,MPN)法[12]和分子生物学检测方法等。平板法是常用的活菌计数方法,操作繁琐、检测周期长、误差大[13],不同分离平板对致病菌计数有一定的影响[14]。MPN法属于半定量检测方法[15-16],为提高检测速度及敏感性,多与聚合酶链式反应(polymerase chain reaction,PCR)方法结合应用[17-18]。平板法和MPN方法在涉及大量样本的情况下需要大量人力[19]。分子生物学检测方法因具有灵敏度高、特异性强、操作简便等优点,在微生物的检测中发挥了巨大的作用[20]。近年来,以real-time PCR为代表的分子生物学技术也逐渐被广泛应用到致病菌的定量检测中。相比较于传统培养法,real-time PCR法快速、简便、灵敏、特异[21],但该方法属于相对定量,结果的准确性和重复性在很大程度上依赖于标准曲线质量及扩增效率,并且需要具备已知浓度的核酸标准品。新兴的微滴数字PCR(droplet digital PCR,ddPCR)被认为是第3代核酸扩增技术[22],通过把稀释到一定浓度的DNA分子分布于10 000~20 000 个微滴中,使大部分微滴中DNA分子的数量为1或0,然后通过PCR扩增和阳性信号的累积读取阳性反应单元数,再根据泊松分布计算出样本中的DNA分子数,从而实现对DNA分子的绝对定量[23-24]。针对现有检测方法的不足,本研究拟采用ddPCR技术建立食品中E. coliO157:H7的快速准确绝对定量方法,为制订食品微生物定量检测标准提供思路和技术支持。
1 材料与方法
1.1 材料与试剂

表1 实验菌株Table 1 List of experimental strains
86 株菌(表1),其中包括4 株E. coliO157:H7标准菌株(CICC 24187、CICC 21530、CICC 10669、FEPAS O157),实验室保存的1 株E. coliO157:H7,1 株产肠毒素(Enterotoxigenic)E. coli标准菌株,1 株肠道致病性(Enteropathogenic)E. coli标准菌株,1 株肠道侵袭性(Enteroinvasive)E. coli标准菌株,1 株肠道集聚性(Enteroaggregative)E. coli标准菌株,2 株其他E. coli标准菌株,55 株实验室分离E. coli,以及18 株其他常见的食源性致病菌标准菌株。所有菌株分别在BHI琼脂平板或2% TSA琼脂平板(活化弧菌)上活化3 代后,挑取单菌落分别接种于10 mL BHI液体培养基或10 mL 3%氯化钠碱性蛋白胨水(弧菌培养)中,37 ℃、160 r/min振荡培养18 h,取1 mL菌液进行10 倍梯度稀释至10-8,用于细菌基因组DNA的提取。
1.2 仪器与设备
培养基 北京陆桥技术股份有限公司;细菌基因组DNA提取试剂盒(货号:DP302) 天根生化科技有限公司;THZ-C-1台式全温振荡器 苏州培英实验设备有限公司;Stomacher 400高效拍打式匀浆器 英国Seward公司;5424小型高速离心机 德国Eppendorf公司;Fr-1io cell恒温培养箱 德国MMM公司;Nanodrop2000超微量分光光度计 美国Thermo公司;QX200 ddPCR系统 美国伯乐公司;7900HT Fast real-time PCR仪、GeneAmp 9700 PCR仪 美国ABI公司。
1.3 方法
1.3.1 引物探针设计、合成与筛选
参考国内外研究[1,25-26],选取E. coliO157:H7具有种属特异性且高度保守的单拷贝溶血素前体蛋白基因hlyA(GenBank EU118026.1)作为靶序列,通过NCBI在线工具进行序列分析和比对,利用Prime Express软件V3.0(ABI, Foster City, CA, USA)设计出引物/探针1 套,序列见表2。探针5’端标记FAM,3’端标记BHQ。
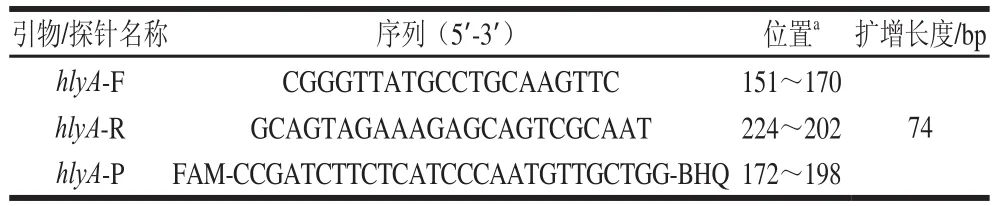
表2 ddPCR引物探针序列Table 2 Primer and probe sequences used for ddPCR
首先,利用该引物/探针通过real-time PCR对表1中所有菌株进行检测,进而通过ddPCR对表1中E. coliO157:H7菌株和其他致泻大肠标准菌株及其他近源菌进行检测,验证该引物探针的有效性和特异性。
1.3.2E. coliO157:H7纯培养液的DNA浓度、平板计数及PCR检测
1.3.2.1 基因组DNA提取与超微量分光光度计测定
E. coliO157:H7 CICC 21530的10 mL BHI过夜培养物(160 r/min、37 ℃摇床振荡培养18 h),取1 mL以磷酸盐缓冲溶液(phosphate buffer saline,PBS)进行10 倍梯度稀释。取过夜培养物原液至10-8稀释度菌液各1 mL采用试剂盒法提取DNA,按照试剂盒说明将核酸溶解在50 μL TE缓冲液中。对提取的纯培养液核酸用超微量分光光度计进行浓度和纯度的测定,确保提取核酸的A260nm/A280nm在1.8~2.0之间,并按下式[19]计算提取E. coliO157:H7基因组DNA浓度:
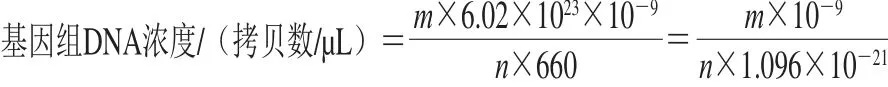
式中:m为超微量分光光度计测得的核酸质量浓度/(ng/μL);n为细菌基因组的长度/bp;根据NCBI上已经发布的E. coliO157:H7基因组的测序数据,其平均长度为5.144×106bp。
1.3.2.2 平板计数
取1.3.2.1节中10 倍梯度稀释的E. coliO157:H7 CICC 21530纯培养液10-6、10-7、10-8稀释度进行平板计数,吸取1 mL各稀释度的菌悬液分别加于2 个无菌平皿内。及时将15~20 mL冷却至46 ℃的平板计数琼脂培养基倾注平皿,并转动平皿使其混合均匀。待琼脂凝固后,36 ℃倒置培养(24±2)h。选取菌落数在30~300 CFU之间、无蔓延菌落生长的平板计数菌落总数,每个稀释度菌落数取两个平板计数结果的平均值。
1.3.2.3 real-time PCR检测
取1.3.2.1节中过夜培养物原液至10-8稀释度菌液各1 mL提取的DNA进行real-time PCR检测,作标准曲线。real-time PCR体系中上下游引物hlyA-F、hlyA-R和探针hlyA-P浓度均为500 nmol/L,然后加入10 μL 2×PCR master mix,DNA模板2 μL,用去离子水补足20 μL。反应体系置于real-time PCR仪中进行扩增。扩增条件为50 ℃酶激活2 min;95 ℃预变性10 min;95 ℃变性15 s;60 ℃退火及延伸1 min,40 个循环。反应完成后利用SDS 3.2软件进行分析。
经超微量分光光度计测得纯培养液核酸浓度,代入1.3.2.1节公式算得提取到的目标菌基因组DNA浓度(拷贝数/μL),根据体积算得试剂盒法提取的50 μL中目标菌基因组浓度(拷贝数/50 μL),也就是1 mL菌液中的浓度(拷贝数/mL),设计引物探针的hlyA基因为单拷贝基因,1拷贝基因组对应1 个CFU菌落,可得到1 mL菌液中的细菌个数(CFU/mL)。
1.3.2.4 ddPCR检测

表3 ddPPCCRR体系Table 3 Composition of ddPCR reaction system
取1.3.2.1节过夜培养物原液至10-8稀释度菌液提取的DNA同时进行ddPCR检测。将20 μL的ddPCR预混液(表3)和70 μL微滴生成油分别加入到8 孔微滴生成卡中,置于微滴生成仪中生成微滴。将生成的油包水的微滴(40 μL)缓慢转移至96 孔板中,封膜后置于PCR仪上进行扩增反应(升降温速率≤2.5 ℃/s):95 ℃预变性10 min;94 ℃变性30 s;55 ℃退火及延伸1 min,45 个循环;98 ℃固化微滴10 min;4 ℃反应结束。将扩增后的96 孔板置于微滴读取仪中,利用QuantaSoft软件进行结果读取和分析。
ddPCR可直接测得每反应的目标基因浓度(拷贝数/反应),即试剂盒法提取的DNA中2 μL的目标基因浓度,计算可得50 μL中的目标基因浓度(拷贝数/50 μL),即1 mL菌液中的目标基因浓度(拷贝数/mL),1拷贝基因对应1 个CFU菌落,也就是1 mL菌液中的细菌个数(CFU/mL)。
1.3.3 人工污染三文鱼样品的平板计数、real-time PCR、ddPCR定量检测
选择GB 4789.36—2016[11]验证无E. coli O157:H7的三文鱼样品作为食品添加基质。取E. coli O157:H7 CICC 21530过夜培养菌悬液的10-3、10-4、10-5、10-6、10-7稀释度菌液各25 mL分别添加到盛有无菌称样25 g三文鱼肉样品的均质袋中,各添加水平分别设3 个样品重复,另无菌称取1 份25 g三文鱼肉样品加入25 mL PBS代替添加菌液作为阴性对照,用拍击式匀浆器拍击2 min,制成系列1∶1含有不同添加水平E. coli O157:H7的污染样品。对以上5 个添加水平的每份人工污染三文鱼样品分别梯度稀释进行平板计数。
取以上5 个添加水平的每份人工污染三文鱼样品匀浆液各2 mL(含1 g三文鱼样品)用试剂盒法提取DNA,最终将核酸溶解在50 μL TE缓冲液中,分别进行real-time PCR和ddPCR检测。real-time PCR以1.3.2.1节梯度稀释的纯培养液DNA作为标准品,以检测Ct值为纵坐标,核酸浓度为横坐标,绘制标准曲线,将人工污染三文鱼样品匀浆液中提取DNA检测Ct值代入回归公式,得到其核酸浓度,代入1.3.2.1节公式得到目标菌基因组DNA的拷贝数(拷贝数/μL),按照1.3.2.3节计算可得2 mL匀浆液中的细菌个数,即1 g三文鱼肉样品中的细菌个数(CFU/g)。
ddPCR直接测得每反应的目标基因拷贝数(拷贝数/反应),按照1.3.2.4节计算可得2 mL匀浆液中的细菌个数,也就是1 g三文鱼肉样品中的细菌个数(CFU/g),从而得到平板计数、real-time PCR及ddPCR 3 种测定方法统一的测定值计算单位,比较平板计数、real-time PCR及ddPCR的测定值效果。
2 结果与分析
2.1 方法特异性分析
real-time PCR方法验证引物和探针的特异性。采用细菌基因组DNA提取试剂盒提取表1中所列菌株纯培养物的基因组DNA,结果表明,7 株E. coli O157:H7菌株(CICC 24187、CICC 10669、实验室自存E. coli O157:H7菌3 株、FEPAS O157、CICC 21530)均出现阳性扩增信号,而其他E. coli及常见的食源性致病菌检测均呈阴性(图1A)。
为了进一步验证ddPCR方法的特异性,梯度稀释7 株E. coli O157:H7菌株(CICC 24187、CICC 10669、实验室自存E. coli O157:H7菌3 株、FEPAS O157、CICC 21530)及8 株近源菌株(Enterotoxigenic E. coli CICC 24190、Enteropathogenic E. coli CICC 24189、Enteroinvasive E. coli CICC 24188、Enteroaggregative E. coli CICC 24186、E. coli CICC 10003、E. coli ATCC 25922、实验室自存E. coli 2 株、S. flexneri ATCC 12022)的各基因组DNA稀释至10-4,用于ddPCR检测,每个样品设2 个平行重复,同时以无菌水代替模板作为阴性对照。结果发现7 株E. coli O157:H7菌株均出现阳性扩增信号,而其他E. coli及常见的食源性致病菌检测均呈阴性,无阳性微滴(图1B),证明该组引物探针在ddPCR扩增体系中具有良好的特异性。
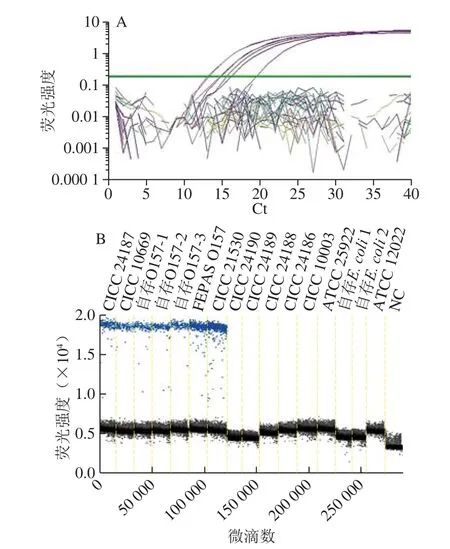
图1 hhllyyAA引物探针real-time PCR(A)和ddPCR(B)特异性结果Fig. 1 Comparison of speci ficity for hlyA between real-time PCR (A)and ddPCR (B)
2.2 E. coli O157:H7 CICC 21530纯培养液的3 种检测方法测定值
2.2.1 real-time PCR测定值
取E. coli O157:H7 CICC 21530过夜培养物1 mL,提取基因组DNA经超微量分光光度计测定质量浓度为671 ng/μL,代入1.3.2.1节公式算得提取到的基因组DNA浓度为1.19×108拷贝数/μL,按照1拷贝基因组对应1 个CFU菌落,则原始菌液菌落数为9.77(lg(CFU/mL))。
2.2.2 平板计数测定值
E. coliO157:H7 CICC 21530过夜培养菌液经10 倍梯度稀释后,进行平板计数,测得原始菌液浓度为2.0×109CFU/mL,即菌落数为9.30(lg(CFU/mL))。
2.2.3 ddPCR测定值及3 种检测方法测定值对比
ddPCR在10-2稀释度达到检测上限,阳性微滴的比率达100%,10-3~10-7稀释度菌液的测定值结果变异系数(coefficient of variation,CV)均小于20%(表4),根据10-3~10-7稀释度菌液的ddPCR结果计算出原菌液中E. coliO157:H7菌落数在8.93~9.11(lg(CFU/mL))范围内(表4)。ddPCR定量限为105 CFU/mL(相当于4.2 拷贝数/反应),检出限为25 CFU/mL(相当于1 拷贝/反应)(图2、表4)。

表4 梯度稀释的E. coollii O157:H7 CICC 21530纯菌液ddPCR定量检测结果Table 4 ddPCR quantitative results of gradient dilutions of pure E. ccoollii O157:H7 CICC 21530 culture

图2 梯度稀释的E. ccoollii O157:H7 CICC 21530纯菌液的ddPCR扩增微滴分布Fig. 2 ddPCR microdroplet distribution of gradient dilutions of pure E. coli O157:H7 CICC 21530 culture

图3 梯度稀释的E. ccoollii O157:H7 CICC 21530纯菌液的real-time PCR扩增图谱Fig. 3 Real-time PCR ampli fication patterns of gradient dilutions of pure E. coli O157:H7 CICC 21530 culture
对各稀释度菌液提取到的DNA同时进行real-time PCR和ddPCR检测,结果发现两者均可检测到10-7稀释度(图2、3)。real-time PCR在10-1~10-7稀释度范围内检测结果呈良好的线性关系(Y=-3.54X+36.91,R2=0.999 8),标准曲线斜率-3.54。ddPCR在测定值范围(10-3~10-7稀释度)内的检测结果具有高度一致性,不同稀释度菌液计算出原菌液中目标菌lg值的CV仅0.9%,平均值为9.01(lg(CFU/mL)),比real-time PCR和平板计数法测定值分别低0.76、0.29(lg(CFU/mL))(图4)。
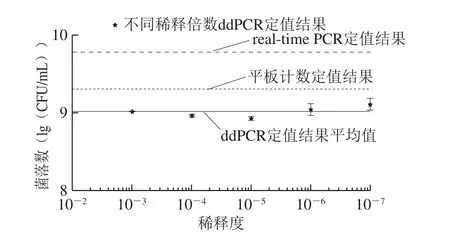
图4 E. ccoollii O157:H7 CICC 21530纯菌液中基因组浓度测定值比较Fig. 4 Comparison of measured genomic DNA concentrations of gradient dilutions of E. coli O157:H7 CICC 21530 culture
2.3 人工污染三文鱼样品的3 种检测方法测定值及分析
2.3.1 平板计数测定值

表5 人工污染三文鱼样品中E. coli O157:H7 CICC 21530的ddPCR、real-time PCR测定值及平板计数结果Table 5 ddPCR, real-time PCR and plate counting results of E. coli O157:H7 CICC 21530 in artifificially contaminated salmon samples
对各个添加水平的人工污染三文鱼样品分别梯度稀释进行平板计数,根据平板计数结果,10-3~10-7菌液污染样品中目标菌E. coliCICC 21530菌落数测定结果分别为1 126 667、85 667、9 000、980、130 CFU/g(表5)。
2.3.2 real-time PCR测定值结果
在进行real-time PCR时,以10 倍梯度稀释的CICC 21530基因组DNA(经超微量分光光度计测定其原液浓度为1.19×108拷贝数/μL)作为标准品,同人工污染样品中提取的DNA一起进行扩增检测。结果发现5 个不同污染水平(10-3~10-7)的三文鱼样品均呈阳性扩增,real-time PCR扩增曲线如图5所示,根据检测Ct值和得到的标准曲线计算污染样品中E. coliO157:H7平均含量分别为4 933 333、613 333、48 667、7 233、575 CFU/g(表5)。
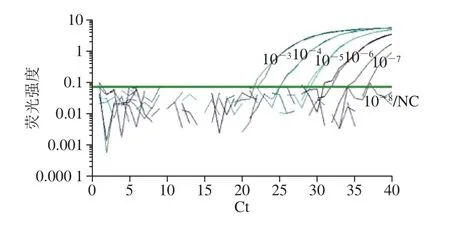
图5 人工污染三文鱼样品real-time PCR检测扩增曲线图Fig. 5 Real-time PCR ampli fication curves of arti ficially contaminated salmon samples
2.3.3 ddPCR测定值结果及3 种定量检测方法比较分析

图6 人工污染三文鱼样品的ddPCR扩增微滴分布Fig. 6 ddPCR microdroplet distribution of arti ficially contaminated salmon samples
图6 显示,随着污染水平的下降,出现的阳性微滴数量逐渐减少,根据阳性微滴数量和泊松分布原理,由QuantaSoft软件测定并根据计算得到E. coliCICC 21530菌落数分别为917 000、80 200、8 330、943、110 CFU/g(表5)。总体而言,real-time PCR和ddPCR的测定结果同平板计数测定值间均存在良好的相关性,但是realtime PCR的测定值结果明显高于ddPCR及平板计数测定值。t检验结果表明ddPCR和平板计数两种方法对稀释度10-4~10-7菌液污染样品的定量结果均无显著性差异(P>0.05,表5);在10-3~10-7菌液污染水平上,realtime PCR的测定结果明显偏高,与ddPCR测定值结果和平板计数均有显著性差异(P<0.05,表5)。可见ddPCR对于人工污染样品的测定结果更加准确,在不经增菌培养的条件下,其检出限为110 CFU/g。
2.4 重复性分析
针对E. coliCICC 21530纯培养液和人工污染三文鱼的ddPCR检测结果均可用来分析整体检测的重复性。由表4和表5可见,在整个定量检测有效动态范围内,针对E. coliCICC 21530hlyA基因的测定结果CV均小于20%,证明该方法用于E. coliCICC 21530定量检测时具有良好的重复性[27]。而且在对人工污染三文鱼样品的检测中,ddPCR定量结果的CV普遍小于real-time PCR,说明ddPCR具有更好的定量重复性,特别是对于低污染水平样品这种优势更加明显(表5)。
3 讨论与结论
国内外已有应用ddPCR技术定量检测致病菌的相关研究,但食品基质多富含蛋白、脂肪、果胶等核酸扩增抑制成分,背景菌构成复杂且浓度较高,因此,对食品基质中食源性致病菌的定量检测研究尚不多见。董莲华等[3]以E. coliO157:H7的rfbE基因为靶基因,建立了可对其准确定量的ddPCR方法。Verhaegen等[28]比较ddPCR及real-time PCR方法定量检测牛粪中产志贺毒素E. coli得出,在不依赖标准品的情况下,ddPCR方法相对realtime PCR在农场绝对定量检测产志贺毒素E. coli中有优势。周巍等[29]以金黄色葡萄球菌nuc基因为靶标基因,建立了发酵乳中ddPCR技术定量检测金黄色葡萄球菌的方法。方佩佩等[30]采用ddPCR技术建立副溶血性弧菌快速定量检测,以鳕鱼为样品进行阳性添加,研究表明ddPCR定量检测技术特异性良好、灵敏度高、结果准确。Gobert等[31]研究表明ddPCR对较低数量的活细胞的定量效果最好,ddPCR在干扰菌背景下,无需建立标准曲线,就可以对少量活菌细胞进行特异性定量。本研究根据高保守特异性序列设计筛选了一套特异性良好的引物探针,利用纯菌液检测建立食品中E. coliO157:H7的ddPCR快速定量检测技术,通过人工污染食品样品的检测验证该方法的实际应用性。通过纯菌液及人工污染样品检测的ddPCR定量结果与平板计数及real-time PCR的定量结果的特异性、灵敏度和重复性比较,认为建立的ddPCR技术可以应用到实际食品的定量检测中。
本研究结果显示平板计数定量结果与ddPCR定量结果接近,real-time PCR定量结果偏高。ddPCR属于绝对定量,具有无需标准物质、无需标准曲线、不受PCR扩增效率影响、对PCR抑制剂不敏感等特点,real-time PCR属于相对定量,结果的准确性和重复性在很大程度上依赖于所绘制的标准曲线质量及扩增效率。目前没有官方权威机构可以提供具有明确量值(即核酸拷贝数或质量浓度)的E. coli O157:H7核酸标准品,本研究采用超微量分光光度计测量核酸分子在260 nm波长处吸光度,根据1.3.2.1节公式计算定量值,建立标准曲线。标准品/样品基质中其他DNA或RNA分子以及蛋白等杂质成分可能会影响测定结果,样品中也可能存在核酸扩增抑制成分导致扩增效率的差异,导致real-time PCR定量结果偏高。
本研究建立的ddPCR检测技术的定量限为105 CFU/mL(相当于4.2 拷贝数/反应),检出限为25 CFU/mL,人工污染食品样品检出限为110 CFU/g,适用于食品中高污染E. coli O157:H7的定量检测应用。现行标准GB 4789.36—2016检测周期为3~5 d,本研究建立的检测方法从提取DNA到ddPCR检测仅需1 d,大幅缩短检测周期,为E. coli O157:H7的防控和监管工作提供技术支持,对食品安全检测、开展风险评估、突发公共卫生事件应急检测、控制疫情扩散、提高病原菌检出效率等均有重要意义[32]。
本研究的目标菌液是经过18 h摇菌培养后细菌活性较好的对数期菌液,但是不能保证菌液中没有死菌体,即游离DNA,ddPCR属于分子检测手段,本实验使用的DNA提取试剂盒并不能有效去除死菌体的DNA,暂时不清楚是否会对实验结果产生决定性影响。选取添加的食品基质是经过筛选的未含有目标菌的三文鱼水产品,但食品类别丰富,基质复杂多样,尤其部分生食食品还可能有多种近源微生物干扰,故提取DNA前有效去除菌液中的游离DNA,进一步降低定量限,扩大检测对象,针对多种食品基质的ddPCR定量检测为以后研究的方向及重点。
猜你喜欢
右江民族医学院学报(2022年2期)2022-05-19
中国土壤与肥料(2021年5期)2021-12-02
河北医学(2021年10期)2021-10-27
青岛大学学报(医学版)(2021年4期)2021-09-10
中国农业科学(2021年6期)2021-03-25
今日农业(2020年20期)2020-11-26
疯狂英语·新悦读(2020年7期)2020-07-30
癌变·畸变·突变(2020年1期)2020-02-12
饮食科学(2016年9期)2016-11-18
健康之路(医药研究)(2015年2期)2015-10-21
