
Autophagy in fate determination of mesenchymal stem cells and bone remodeling
2020-09-18 13:28
World Journal of Stem Cells 2020年8期
Xiao-Dan Chen,Jia-Li Tan,Yi Feng,Li-Jia Huang,Mei Zhang,Bin Cheng,Hospital of Stomatology,Sun Yat-sen University;Guangdong Provincial Key Laboratory of Stomatology;Guanghua School of Stomatology,Sun Yat-sen University,Guangzhou 510055,Guangdong Province,China
Abstract
Key words:Mesenchymal stem cells;Autophagy;Cell self-renewal;Cell differentiation;Cytoprotection;Bone remodeling
INTRODUCTION
Mesenchymal stem cells (MSCs) are a heterogeneous cellular population that can be detected in and isolated from bone marrow,adipose,vascular,umbilical cord,placenta,skin,and kidney[1-3].Characterized by their potential of self-renewal and differentiation into osteogenic,chondrogenic,and adipogenic lineages,they are considered promising therapeutic agents that confer a positive benefit to bone maintenance,repair,and regeneration[4].Therefore,a thorough understanding of the underlying mechanisms regulating MSCs function would offer great promise in the field of bone regenerative medicine.
Autophagy is a conserved degradation process during which proteins and damaged organelles are engulfed by autophagosomes and then fused with lysosomes to be degraded for intracellular recycling to fuel cellular renovation[5,6].There are three types of autophagy in mammals,including macroautophagy[7],microautophagy[8],and chaperone-mediated autophagy[9],among which macroautophagy is in the spotlight for its crucial effects on cell biology and will be henceforth referred to as “autophagy”in this review.
Recent evidence has shed light on a fundamental role of autophagy in the fate determination of MSCs and the maintenance of bone homeostasis.In addition,autophagy has also been implicated as an immediately available cytoprotective mechanism in MSCs against stress[10,11].Dysfunction of autophagy would impair the function of MSCs,leading to imbalances of bone remodeling and thus inducing a wide range of aging and degenerative bone diseases.Further delineation of the relationships among autophagy,MSCs function,and bone homeostasis would uncover new avenues for novel therapeutic strategies for bone repair and regeneration.
REGULATORY MECHANISMS OF AUTOPHAGY
Autophagy is regulated by a number of signaling pathways,among which the most well-known are the adenosine monophosphate-activated protein kinase (AMPK) and the phosphoinositide3 kinase (PI3K)/AKT pathways,which converge on mammalian target of rapamycin (mTOR),a well-recognized negative regulator of autophagy that integrates nutrient signals[12](Figure1).mTOR recruits other regulatory proteins to form two distinct complexes,mTORC1 and mTORC2,and mTORC1 is involved in autophagy regulation[13].
AMPK is a principal intracellular energy sensor,which conserves energy by inhibiting mTOR[14]by phosphorylating and potentiating tuberous sclerosis complex(TSC) or by directly binding to RAPTOR,a key subunit of mTORC1[15],and consequently inducing autophagy[16].Furthermore,the PI3K/AKT pathway is also an important mTOR modulator that inhibits the mTOR repressor TSC[17],activates mTOR,and then blocks autophagy activity[12].In addition,wnt/β-catenin has been shown to be a negative regulator of autophagy,while PTEN induces autophagy by inhibiting the PI3K/AKT/mTOR pathway,and activated EGFR/Ras/MEK/ERK,JUK/c-Jun,and p38 MAPK signaling pathways have also been revealed as stimulators of autophagy[18].
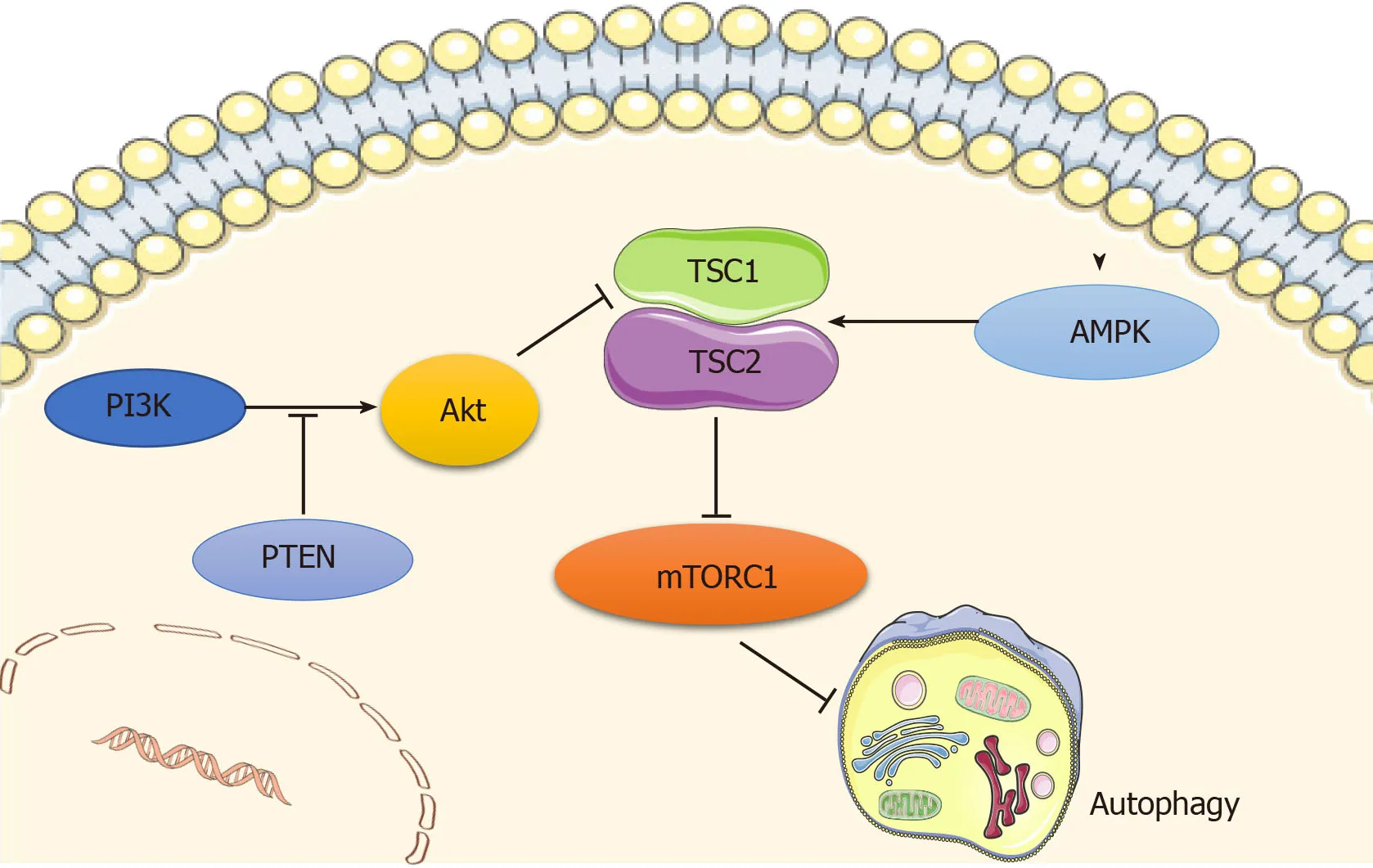
Figure1 Main signaling pathways regulating autophagy.Autophagy is regulated by the adenosine monophosphate-activated protein kinase (AMPK) and PI3K/AKT pathways,which converge at mammalian target of rapamycin (mTOR) that functions as a negative regulator of autophagy.AMPK inhibits mTOR by phosphorylating tuberous sclerosis complex (TSC),and consequently inducing autophagy.PI3K/AKT pathway inactivates TSC,phosphorylates mTOR,and then blocks autophagy activity,while PTEN acts as a brake upstream of Akt.Original elements used in this diagram are from Servier Medical Art (http://smart.servier.com/).AMPK:Adenosine monophosphate-activated protein kinase;mTOR:Mammalian target of rapamycin;TSC:Tuberous sclerosis complex.
AUTOPHAGY AND MSCS FUNCTION
Autophagy and the lineage determination of MSCs
Considerable evidence has shown a pivotal regulatory role of autophagy in self–renewal capacity and lineage determination of MSCs.Induction of autophagy in bone mesenchymal stem cells (BMSCs) may account for a decrease in their S-phase population and trigger their differentiation into neurons[19].Despite some controversy,Isomotoet al[20]clarified that rapamycin does not have a spontaneous osteogenic effect on MSCs,while most studies have confirmed that autophagy contributes to the switch between osteogenesis and adipogenesis of BMSCs.More specifically,MSCs tend to accumulate undergraded autophagic vacuoles and undergo little autophagic turnover,while osteogenic differentiation of MSCs results in more autophagic turnover[21].Induction of osteogenic differentiation of human gingiva-derived MSCs (HGMSCs)potentiates autophagy signaling,while inhibition of autophagy precludes osteoblast differentiation of HGMSCs[22].The autophagy inducer rapamycin promotes osteoblast differentiation in human embryonic stem cells (ESC) by interfering with mTOR while augmenting the BMP/Smad signaling pathway[23].
Osterix-expressing cells with a specific deletion ofTSC1,a positive regulator of autophagy,have shown thatTSC1deficiency is responsible for the reduction of bone mass,as characterized by inhibition of osteogenesis,enhancement of osteoclastogenesis,and elevation of bone marrow adiposity[24].Consistently,TSC1deficiency in BMSCs results in decreased proliferation and a tendency to differentiate into adipocytes instead of osteoblasts[24].
Other studies have provided evidence that early mTOR suppression accompanied by late Akt/mTOR activation contributes to osteoblast differentiation of MSCs[25].Accordingly,activation of autophagy by mTOR inhibition facilitates osteoblast differentiation[25],though whether late mTOR induction and subsequent autophagy inhibition would stimulate or interfere with osteogenesis remains to be elucidated.Another study in MC3T3 cells also revealed that early activation and subsequent inhibition of AMPK are indispensable for osteoblast differentiation[26].Given that mTOR functions as an inhibitor and AMPK as a stimulator of autophagy,these two studies coincide in that autophagy is fueled at first and then abrogated during osteoblast differentiation.We theorized that such time-dependent catabolic dynamics seem fundamental to ensure the ever-changing energy demands during all stages of osteogenesis.
Autophagy in aging/senescence of MSCs
Although several studies have revealed that autophagy is activated during aging in cells such as fibroblasts[27]and BMSCs[28],the mainstream view currently is that with aging,autophagy decreases in different kinds of tissues,ranging from the kidney to the brain[29,30].Indeed,it has been reported that autophagy activity is significantly reduced in aged BMSCs compared with their young counterparts[31].Basal autophagy has a crucial role in the maintenance of the young state of satellite cells,and dysfunction of autophagy leads to cell senescence as indicated by the decrease in satellite cell number and function[32].In addition,blockage of autophagy converts young BMSCs to a relatively aged state by impairing their osteoblast differentiation and proliferation potential while promoting their adipocyte differentiation ability.Correspondingly,activation of autophagy turns aged BMSCs into a young state by strengthening osteoblast differentiation and proliferation potential while impairing adipocyte differentiation capacity[31].Likewise,pretreatment with rapamycin remarkably alleviates MSC aging induced by D-gal and decreases of p-JNK,p-38,and ROS generation,supporting the concept that autophagy exerts a protective role in MSCs senescence[33].This protective effect of rapamycin on MSCs senescence can be abolished by increasing the ROS level,and inhibition of p38 can rescue the H2O2-induced MSCs senescence,which suggests that ROS/JNK/p38 signaling contributes to mediating autophagy-delayed MSCs senescence[33].
Collectively,autophagy is a surveillance pathway that tightly controls fate decisions of MSCs,and therefore it should be considered when searching for methods to maintain the pluripotency of MSCs.
CYTOPROTECTION OF AUTOPHAGY IN MSCS UNDER STRESS
Hypoxic conditions
Autophagy is known to exert cytoprotection for MSCs under stress conditions[34].It has been demonstrated that hypoxia-pretreated MSCs exhibit AMPK/mTOR signaling activation,autophagy enhancement,and pro-angiogenic effect improvements[35].Similarly,Zhanget al[36]showed that the autophagy inhibitor 3-methyladenine (3-MA)promotes hypoxia-induced apoptosis,while a positive inducer of autophagy,rapamycin,decreases hypoxia-induced apoptosis,suggesting that autophagy seems to be a protective element in MSCs under hypoxic stress and that atorvastatin could improve BMSCs survival during hypoxia by enhancing autophagyviathe AMPK/mTOR pathway.However,there are also studies showing that hypoxia activates the autophagic flux of BMSCs through the AMPK/mTOR pathway and that activation of the latter process plays an important role in hypoxia-induced apoptosis[37-39].This complicated scenario might be due to the heterogeneity and sitespecific properties of the MSCs.For instance,BMSCs derived from the mandible have higher expression of the stemness markersNanog,Oct-4,andSox2,as well as stronger autophagy and anti-aging capacities under normoxia or hypoxia,when compared to those derived from the tibia[40].
Oxidative stress
A recent study showed that oxidative stress-induced MSCs death could be prevented by carbon monoxide,and this protective effect is due to an increase of autophagy[41].Autophagy facilitates the turnover of damaged cellular components,which may result in improved cellular survival in the setting of oxidative injury.Therefore,depletion of autophagy in MSCs exacerbates oxidative stress-induced MSCs death[41].Augmenting autophagy by JNK activation also protects MSCs against oxidative damage,thereby improving MSCs survival[42].Preconditioning or coconditioning with rapamycin alleviates,while 3-MA aggravates,H2O2-induced cell apoptosis[34].Likewise,H2O2-treated human MSCs (hMSCs) activatesFOXO3and then induces autophagy in response to the elevated ROS level,thus preventing oxidative injury.In line with this,suppression of autophagy impairs ROS elimination and the osteogenic capacity of hMSCs[43].However,it is worth noting that these cytoprotective effects of autophagy on MSCs in the context of oxidative damage seem to act in a stress severity- and duration-dependent manner.Autophagy flux is considered to be a self-defensive process during the early stage of MSCs injury induced by H2O2,and this protective effect would be abolished after sustained oxidative exposure (i.e.,6 h),as demonstrated by increased levels of caspase-3 and caspase-6[34],which indicates that adaptive autophagy contributes to an improved survival rate of MSCs under stress,while destructive autophagy is induced when it fails to manage excessive stress[44].
Irradiation stress
As the main mechanism by which cells initiate self-protection in a radiation microenvironment[45,46],autophagy triggers a DNA damage response by regulating DNA repair and checkpoint protein levels[47].Some studies have reported that autophagy decreases after irradiation,suggesting an impairment in eliminating damaged cellular components[48].Activation of autophagy in MSCs reduces radiationgenerated ROS and DNA damage,leading to the maintenance of stemness and differentiation potential[34,49],while suppression of autophagy results in more ROS generation,DNA damage,and worsening of self-renewal ability[49].This radioprotective role of autophagy on MSCs is further supported by the observation that hypoxia increases both the autophagy level and MSCs radioresistanceviaERK1/2 and mTOR signaling[50-52],suggesting a positive relationship between autophagy and the radioresistance of MSCs.
Inflammatory stress
Increasing evidence has shown that autophagy provides a crucial line of induction and modulation of the inflammatory status of MSCs.In a TNF-α/cycloheximide-induced inflammatory environment,enhancement of autophagy reverses the decreased survival rate of MSCs,while inhibition of autophagy aggravates apoptotic progression[53].Nevertheless,there have been reports of the adverse regulatory effects of autophagy in MSCs.The inflammatory cytokines TNF-α and IFN-γ synergistically enhance autophagy in MSCs,as evidenced by increased expression ofBECN-1/Beclin-1.Knockdown ofBeclin1improves the therapeutic effects of MSCs and increases their survival by promotingBcl-2expressionviathe ROS/MAPK1/3 pathway[54,55].Wanget al[56]showed that autophagy is triggered in MSCs in response to a liver fibrosis(LF) microenvironment.Of note,autophagy suppression can improve the antifibrotic potential of MSCs and this contributes to their inhibitory effects on T lymphocyte infiltration as well as the production of inflammatory cytokines TNF-α and IFN-γ[56].Additionally,inhibition of autophagy increases ROS accumulation and MAPK 1/3 activation in MSCs,which are essential for prostaglandin E2 expression to exert an immunoregulatory function,thus resulting in enhanced suppression upon activation and expansion of CD4+ T cells and leading to upregulation of the immunosuppressive function of MSCs[54].This implies that autophagy may not always be beneficial in protecting the reparative effect of MSCs.Hence,modulating the multifaceted effects of autophagy in MSCs would provide a novel strategy to improve MSCs-based therapy.
AUTOPHAGY AND BONE REMODELING
Bone remodeling is dynamic process that helps to maintain bone integrity and mineral homeostasis.There are three kinds of cell types involved in bone remodeling:Osteoclasts,osteoblasts,and osteocytes[57].Among them,both osteoblasts and osteocytes are derived from BMSCs,while osteoclasts have a hematopoietic origin[58].Osteoclasts are multinucleated cells that initiate bone remodeling by digesting old bone,whereas osteoblasts are responsible for synthesizing and secreting bone matrix to form new bone[58].Osteocytes,as the most abundant cell type in bone tissue,are pivotal in bone remodeling by coupling osteoblasts and osteoclasts activities[59]viathe receptor activator of NF-kappa B (RANK)/receptor activator of NF-kappa B ligand(RANKL)/osteoprotegerin (OPG) system[57].Though still in its infancy,growing evidence has clarified that autophagy is closely related to bone remodeling mediated by osteoclasts,osteoblasts,and osteocytes,by which it exerts a critical role in coupling bone formation and bone resorption,thus maintaining normal postnatal bone homeostasis[60].
Autophagy in osteoclasts
Previous research has demonstrated that activation of autophagy by AMPK signaling inhibits osteoclast differentiation[61].Moreover,autophagy induced by OPG attenuates osteoclast bone resorptionviathe AKT/mTOR/ULK1 axis[62].Similarly,autophagy favors OPG-mediated inhibition of osteoclast differentiation and bone resorption through the AMPK/mTOR/p70S6K signaling pathway[63].These data highlight a negative regulation of autophagy in osteoclastogenesis.However,Caoet al[64]showed that inhibiting autophagy suppresses TRPV4-induced osteoclast differentiation and osteoporosis via the Ca2+-calcinertin-NFATc1 pathway.In addition,JNK1-induced autophagy decreases apoptosis of osteoclast progenitors and stimulates RANKLmediated osteoclastogenesis[65],which shows a positive effect of autophagy on osteoclast activity,suggesting a potential role of autophagy in initiating bone remodeling.
Autophagy in osteoblasts
Moreover,autophagy promoted by estradiol protects osteoblasts from apoptosisviathe ER-ERK-mTOR axis[66].Interestingly,both early proliferation and differentiation are not interfered by inactivation of autophagy byFIP200ablation,a fundamental element of mammalian autophagy,while osteoblast terminal differentiation is adversely affected,as shown by defective nodule formation[67],which suggests a positive role of autophagy in nodule formation.Consistently,osteoblastic mineralization is found to be accompanied by activation of autophagy,in which vacuoles could act as vehicles for crystals secretion.Thus,osteoblast specific autophagy deficient mice exhibit a significant reduction of mineralization and bone mass[68].Bone mass in osteoblast-specificAtg7conditional knockout (cKO) mice is significantly decreased compared with the control,the phenotype of which is caused by a decrease of osteoblast number and mineralization,as well as an increase of osteoclast number and osteoclast activity[60].These results mean that autophagy exerts a critical role in osteoblast differentiation.
Autophagy in osteocytes
Yanget al[69]suggested a negative correlation between osteocyte autophagy and an ovariectomy (OVX) induced oxidative stress condition and bone loss.Reduction of autophagy by estrogen deficiency promotes the apoptosis of osteocytes,whereas restoration of autophagy strengthens the anti-apoptotic effects to improve osteocyte viability[70].Osteocytes-specific cKO ofAtg7,a key gene involved in autophagy,results in reduced bone mass,decreased cancellous and cortical bone thickness,and increased cortical bone porosity at 6 mo for both male and female mice,which contributes to decreases in osteoblast number,bone formation rate,and osteoclast number[71].In addition,EphrinB2 in osteocytes limits autophagy to ensure bone quality by controlling mineral accumulation,while dysfunction of the osteocytic EphrinB2-autophagy signal results in bone fragility[72].These findings emphasize a central role of autophagy in regulating osteocyte biology as well as bone remodeling.
AUTOPHAGY AND BONE DISEASE
Increasing numbers of studies have revealed a crucial role of autophagy in the development and progression of many kinds of bone disease,such as osteopetrosis,Paget's disease,and osteoporosis[73-75].Recently,a genome-wide association study of wrist bone mineral density caught our attention since it revealed a close relationship between osteoporosis and autophagy[76].Further research demonstrated that MSCs from an osteoporosis mouse model induced by estrogen deficiency exhibit reduced autophagy,which is associated with abnormal regenerative function[73].Interestingly,restoration of autophagy by administrating rapamycin rescues the regenerative function of MSCs and protects OVX mice from osteoporotic development[73].A similar decreased level of autophagy is also observed in OVX rats,while restoration of autophagy in osteoblasts by overexpressing autophagy gene damage-regulated autophagy modulator(DRAM)inhibits osteoblast proliferation and promotes their apoptosis[77].In addition,activation of autophagy restored bone loss in aged mice[31],whereas blockade of autophagy alleviated glucocorticoid-induced and OVX-induced bone loss by interfering with osteoclastogenesis[75].What’s more,defective autophagy in osteoblasts results in mouse osteopenia[67].In experimental models of arthritis,rapamycin treatment can reduce the number of osteoclasts and osteoclast formation,thus inhibiting bone absorption in young rats[78].Furthermore,rapamycin reduces bone resorption in renal transplant patients[79],enhances osteogenic differentiation in a mouse model of osteopenia[80,81],and ameliorates age-induced bone defects in aged rats[82].Further study is warranted to explore the potential application of autophagy modulators as preventive or therapeutic strategies in bone disease.
CONCLUSION AND FUTURE PERSPECTIVES
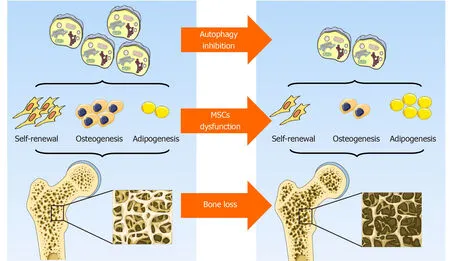
Figure2 Role of autophagy in mesenchymal stem cells integrity and bone homeostasis.Autophagy contributes to the maintenance of mesenchymal stem cells integrity by preserving their self-renewal and osteoblast differentiation potential while inhibiting adipocyte differentiation,thus orchestrating bone homeostasis.Original elements used in this diagram are from Servier Medical Art (http://smart.servier.com/).MSCs:Mesenchymal stem cells.
In general,although the prevailing views currently support the hypothesis that autophagy contributes to the maintenance of MSCs integrity by preserving their selfrenewal and osteoblast differentiation potential while inhibiting adipocyte differentiation,thus orchestrating bone homeostasis (Figure2),some data are still somewhat controversial.To some extent,autophagy is a dynamic process that depends on immediate cellular energy demands.Thus,it is necessary to investigate its biological role along a timeline instead of at a single isolated time point.In addition,autophagy may act as a double-edged sword,the effects of which are modified in response to the features,severity,and duration of a specific stress.Furthermore,the latest study emphasizes a critical role of mitochondrial autophagy,or mitophagy,in stem cell fate plasticity and determination[83,84].Effects of an underlying crosstalk between autophagy and endoplasmic reticulum stress in MSCs and bone biology regulation is also beginning to be uncovered[85].Further study is needed to lift the veil on the pleiotropy of autophagy,its reciprocal and functional interactions with other organelles,and their role in MSCs functional orchestration and bone biology modulation.
 World Journal of Stem Cells2020年8期
World Journal of Stem Cells2020年8期
- World Journal of Stem Cells的其它文章
- Hunting down the dominating subclone of cancer stem cells as a potential new therapeutic target in multiple myeloma:An artificial intelligence perspective
- Role of mesenchymal stem cell derived extracellular vesicles in autoimmunity:A systematic review
- Human embryonic stem cell-derived mesenchymal stem cells improved premature ovarian failure
- Assessment of tobacco heating system 2.4 on osteogenic differentiation of mesenchymal stem cells and primary human osteoblasts compared to conventional cigarettes
- Mesenchymal stem cell-derived exosomes:Toward cell-free therapeutic strategies in regenerative medicine
- Human embryonic stem cells as an in vitro model for studying developmental origins of type 2 diabetes