
月桂烯的活性阴离子聚合及其“巯基-双键”点击反应
2020-09-20 11:35马红卫
合成树脂及塑料 2020年4期
黄 帅,韩 丽,马红卫,李 杨
(大连理工大学 精细化工国家重点实验室,辽宁省大连市 116024)
随着石油资源的大量消耗,生物资源和可再生资源成为人们关注的热点。月桂烯(Mc)作为一种资源丰富的单萜烯化合物,广泛存在于多种植物中[1-2],其结构类似于异戊二烯和丁二烯,可作为来源于石油的共轭二烯类化合物的替代物[3-4]。基于阴离子聚合方法合成的橡胶和热塑性弹性体具有相对分子质量高、相对分子质量可控及分布窄的特点,并且具有优异的力学性能[5]。均聚月桂烯(PM)单元上含有多种类型的双键结构,可进行不同程度的后功能化,实现新型功能化及绿色生物基橡胶和弹性体的制备。因此,基于Mc的活性阴离子聚合及其功能化研究具有重要意义[6-7]。近年来,基于通用高分子材料进行后功能化改性,实现新型高性能材料的合成得到了广泛的发展。“巯基-双键”点击反应具有高效、高选择性等优点,适用于多种聚烯烃及巯基类衍生化合物的改性研究,得到了众多研究者的青睐[6-9]。Hong Miao等[10]研究了不同类型双键(包括末端双键、环上双键、中间双键)与巯基化合物进行“巯基-双键”点击反应的活性;Matic等[7]研究了不同结构巯基类衍生化合物与双键的点击反应活性;Uygun等[11]研究了引发剂类型、反应时间对“巯基-双键”点击反应效率的影响。与硅氢加成、酯化和醚化等反应相比,“巯基-双键”点击反应对材料的功能化具有非常好的效果,能够实现材料无金属、无卤素等优点。综上所述,双键类型、巯基化合物结构、引发剂类型、反应时间等均会影响“巯基-双键”点击反应,目前所报道的“巯基-双键”点击反应大多基于双取代双键的点击研究,三取代双键的“巯基-双键”点击反应研究较少。本课题组长期致力于活性阴离子聚合的研究,基于阴离子聚合合成的聚合物具有相对分子质量可控且分布窄等特点[12-15],为可控功能化研究提供了保证。研究表明,采用活性阴离子聚合方法合成的PM以三取代双键结构为主,少量双取代双键结构[5,7],为研究三取代双键的“巯基-双键”点击反应提供了条件。本工作采用活性阴离子聚合方法合成了PM、苯乙烯(St)-Mc两嵌段共聚物(SM),以及St-Mc-St三嵌段共聚物(SMS)。在365 nm紫外光辐照下,分别以2,2-二甲氧基-2-苯基苯乙酮(DMPA)与二苯甲酮(BP)为光引发剂,研究3-巯基丙酸甲酯与PM的“巯基-双键”点击反应,探索了光引发剂类型、反应时间和投料比对点击反应的影响,采用凝胶渗透色谱(GPC)和核磁共振氢谱(1H-NMR)对聚合物的相对分子质量和点击效率进行了研究。
1 实验部分
1.1 主要原料
Mc,St:纯度均为95%(w),东京化成工业株式会社。在氩气保护下,将Mc与St置于密封的圆底烧瓶中,用CaH2搅拌72 h,真空蒸馏保存,使用前用二丁基镁搅拌5 h,在真空系统下蒸馏,置于装有Na屑的密封瓶中保存。极性调节剂N,N,N,N-四甲基乙二胺(TMEDA)采用文献[16]的方法精制,然后在10.08%(w)的苯溶剂中稀释。甲苯,环己烷:经Mbraun SPS-800型溶剂处理系统纯化后装入密封瓶,置于手套箱中保存。仲丁基锂(sec-BuLi),实验室合成,用苯溶剂稀释到0.406 8 mol/L后置于手套箱中保存。异丙醇,通过氩气鼓泡脱除氧气,密封保存。3-巯基丙酸甲酯,纯度98%(w),美国Sigma-Aldrich公司。DMPA,BP:纯度均为98%(w),美国Alfa Aesar公司,直接使用。
1.2 活性阴离子聚合与“巯基-双键”点击反应
1.2.1 活性阴离子聚合
PM的合成:为了保证引发过程一致,在手套箱中进行活性阴离子聚合,Mc单体与环己烷溶剂的体积比为1∶10。用定量密封注射器向安瓿瓶中加入单体Mc(5 g,0.037 mol)与环己烷(63 mL) ,根据预先设计的数均分子量(Mn),迅速向上述体系中定量加入0.406 8 mol/L的引发剂sec-BuLi,将反应体系密封,置于40 ℃水浴中反应5 h后,迅速加入1 mL终止剂异丙醇,将反应液倒入大量甲醇溶剂中,沉降后将试样真空干燥48 h至恒重,得到纯PM(Mn为2.5×103,5.0×103,10.0×103的PM分别记作PM-1,PM-2,PM-3)。PM的合成反应示意见式(1)。
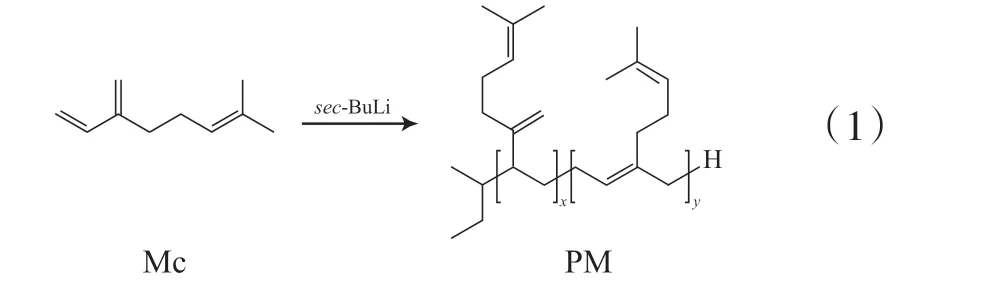
SM的合成:分别根据顺序投料、一步投料(同时添加极性调节剂)的活性阴离子聚合方法合成。
顺序投料法:根据设计的目标相对分子质量,即SM中St与Mc的相对分子质量均为5 000(记作St-5k-Mc-5k)或者St与Mc的相对分子质量分别为5 000,10 000(记作St-5k-Mc-10k)。在苯溶剂(10 mL)中,先用密封注射器顺序加入引发剂sec-BuLi (0.49 mL,0.2 mmol),再迅速加入St(1 g,0.01 mol),在安瓿瓶中不断搅拌5 h,获得转化率接近100%的红色溶液聚苯乙烯(PS)活性种PS-Li(取少量PS-Li用异丙醇终止,作为嵌段共聚物的前驱体)。同步,将定量苯溶剂加入安瓿瓶中,再加入定量Mc单体,将活性PS-Li加入到Mc的苯溶液中,Mc迅速被引发,溶液变成黄色,将安瓿瓶密封,于40 ℃水浴中反应5 h,用异丙醇终止反应。
一步投料法:在定量干燥的苯溶剂中,同时加入St(1 g,0.01 mol)和定量Mc单体,然后加入引发剂sec-BuLi (0.49 mL,0.2 mmol),再迅速加入极性调节剂TMEDA (0.23 g,0.2 mol),n(sec-BuLi)∶n(TMEDA)=1∶1,将安瓿瓶密封放入35 ℃水浴中反应5 h,用异丙醇终止反应。
将上述溶液倒入大量的甲醇中沉降,静置24 h后过滤,产物真空干燥24 h以上至恒重,得到SM。
SMS的制备与上述方法类似,采用顺序投料法合成。
SM与SMS的合成反应示意见式(2)~式(4)。
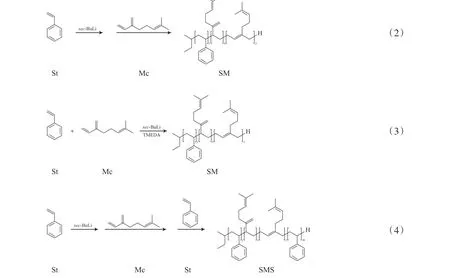
1.2.2 “巯基-双键”点击反应
将PM-2(100 mg)与甲苯(60 mL) 置于圆底烧瓶中,充分搅拌至溶解,分别加入3-巯基丙酸甲酯与光引发剂 (DMPA或BP),在氩气保护下,经过冷冻—脱气—冷冻三次循环操作后,充分排除体系内的氧气对实验的影响,在365 nm紫外光辐照下,搅拌反应12~48 h,为避免紫外光光源散失,反应装置用锡箔纸包裹后置于暗箱中,反应结束后,将反应液倒入大量甲醇溶液中,以除净未反应的3-巯基丙酸甲酯单体,将试样真空干燥48 h至恒重,得到3-巯基丙酸甲酯接枝PM(PM-SH)。PM与3-巯基丙酸甲酯的点击反应合成路线见式(5)。
1.3 结构表征
聚合物的Mn及相对分子质量分布(Mw/Mn,Mw为重均分子量)采用美国Waters公司的Waters 1515型和英国Malvern公司的TDA-305型凝胶渗透色谱仪测定。Waters 1515型凝胶渗透色谱仪配备2414 RI型检测器和两根分离柱(HT4和HT5),流动相为四氢呋喃(THF)(1 mL/min,35 ℃),不同相对分子质量PS为标样;TDA-305型凝胶渗透色谱仪配备多重检测器 (RI:示差检测器,VISC:黏度检测器;LS:光散射检测器)和两根分离柱(T6000 M×2),流动相为THF(1 mL/min,35 ℃)。1H-NMR采用德国布鲁克公司的400 MHz型核磁共振波谱仪测定,氘代氯仿为溶剂,四甲基硅烷为内标。
2 结果与讨论
2.1 Mc的活性阴离子聚合
从图1可以看出:不同MnPM的GPC谱线均呈现单峰、窄分布的特点,且随着PM的Mn设计值的增大,流出曲线的保留时间明显减少,符合活性阴离子聚合的特征。
从图2可以看出:化学位移(δ)为4.6~5.0是PM的3,4-双键的两个氢(b积分区域),δ为5.0~5.2是PM的1,4-双键的两个氢及3,4-双键的一个氢(a积分区域)。
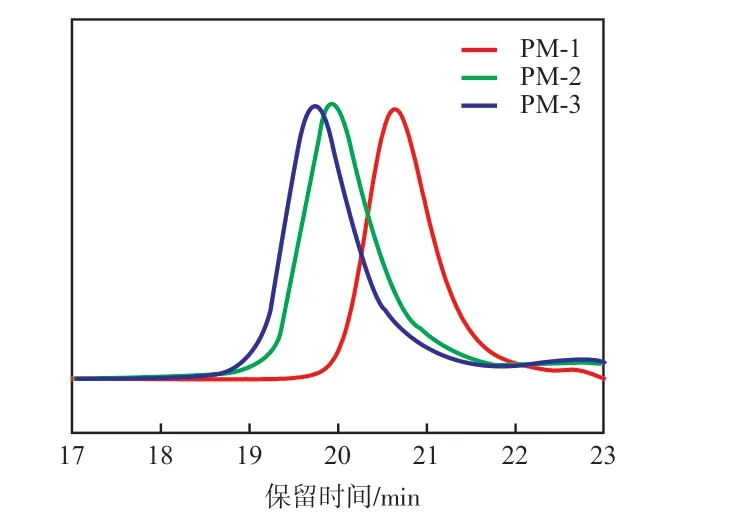
图1 不同相对分子质量PM的GPC曲线Fig.1 GPC curves of PM with different relative molecular weights

图2 PM的结构式及1H-NMRFig.2 Structure and 1H-NMR of PM
从表1可以看出:PM的3,4-双键摩尔分数接近7%,1,4-双键摩尔分数接近93%,说明在没有极性调节剂的条件下,Mc的活性阴离子聚合选择性以1,4-聚合为主。从表1还可以看出:配置多重检测器凝胶渗透色谱仪测试的Mw明显大于配置单一RI检测器凝胶渗透色谱仪测试的Mw,并且配备多重检测器的GPC测试得到的Mw更接近于聚合物真实的Mw。以上结果的差异主要是由于两种测试方法的原理不同。对于配置单一RI检测器的凝胶渗透色谱仪,其色谱柱需要采用不同相对分子质量的PS标样进行校正,获得不同相对分子质量PS的标准曲线,根据所获得的聚合物在GPC中流出曲线的保留时间,基于PS标准曲线,确定试样的Mn,而此时获得的Mn是所合成聚合物相对于PS标样的相对分子质量。近年来,光散射的发展确定了最准确的相对分子质量测定方法。对于配置多重检测器的凝胶渗透色谱仪主要包括LS检测器和RI检测器等。根据光散射等式Rθ=Mw·c·(dn/dc)2(其中,Rθ为瑞利系数;c为低角度激光散射的LS响应常数;dn/dc为折光指数增量,通过多重检测器测试得到,说明在特定的聚合物浓度下,dn/dc决定了聚合物Mw的准确性。RI检测器将强度信号与浓度信号转换,从而通过多重检测器凝胶渗透色谱仪获得了准确的Mw和dn/dc。综上所述,当所合成的聚合物与PS标样结构相似时,配置RI检测器的凝胶渗透色谱仪可以获得相对准确的Mn。反之,当聚合物与PS标样结构相差较大时,配置单一RI检测器的凝胶渗透色谱仪很难获得准确的Mn。此时,配置LS检测器的凝胶渗透色谱仪可以获得准确的Mw,因此,配备多重检测器的凝胶渗透色谱仪测试得到的Mw更接近聚合物真实的Mw。
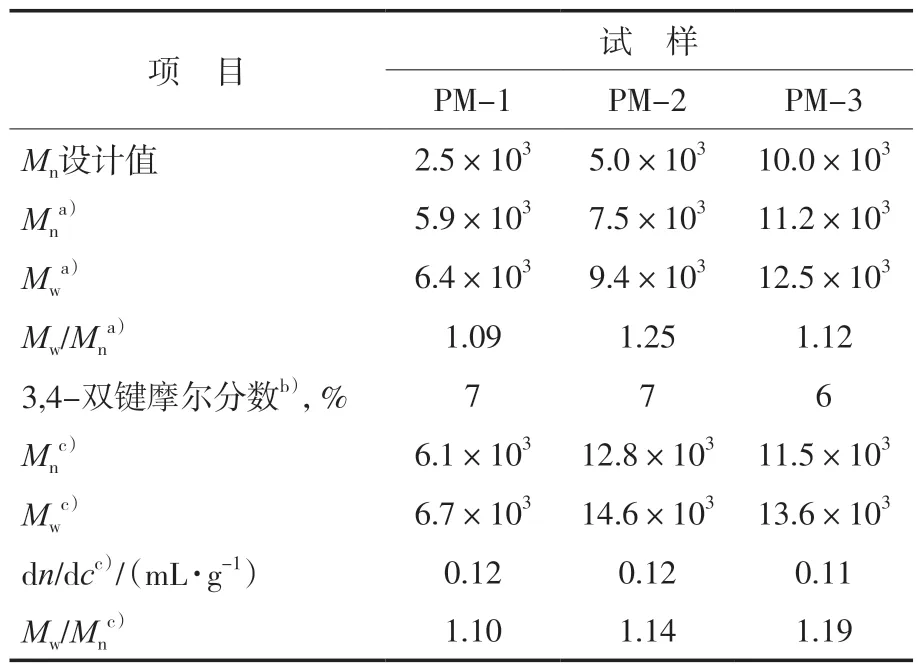
表1 PM的分子设计及分子组成Tab.1 Molecular design and molecular composition of PM
从图3可以看出:δ为4.6~5.0是PM的3,4-双键的两个氢(c积分区域),δ为5.0~5.2是PM的1,4-双键的两个氢及3,4-双键的一个氢(b积分区域),将PS嵌段(a积分区域)的特征峰归属积分面积固定,当加入极性调节剂TMEDA后,3,4-双键的谱峰面积显著提高,而1,4-双键的谱峰面积明显降低。
根据1H-NMR特征峰归属和公式计算,可以获得PM嵌段的微观结构。从表2可以看出:不加入极性调节剂TMEDA的情况下,Mc的活性阴离子聚合仍然以1,4-聚合为主,3,4-双键和1,4-双键比例与均聚物结构接近。当加入极性调节剂TMEDA后,Mc的双键活性发生了改变,其活性阴离子聚合选择性也发生了变化,由1,4-聚合为主转变为1,2-聚合为主,3,4-双键摩尔分数达64%,同时,通过一步投料法添加极性调节剂TMEDA[n(sec-BuLi)∶n(TMEDA)=1∶1],得到的SM经计算表明,其结构仍为嵌段结构。进一步通过表征发现,SMS可通过阴离子顺序加料法制备,制备的嵌段共聚物相对分子质量分布窄,且嵌段相对分子质量可实现精准调控。

图3 添加调节剂前后SM的1H-NMRFig.3 1H NMR of SM before and after adding modifier

表2 SM与SMS的GPC及1H-NMR表征数据Tab.2 GPC and 1H-NMR data of SM and SMS
从表2可以看出:不添加TMEDA,聚合物的相对分子质量更接近设计值;同时通过中间取样的方式得到顺序投料的PS段GPC曲线,从图4可以看出:GPC曲线明显向左偏移,表明成功合成了嵌段共聚物SM。进一步对比表2数据发现,在不添加极性调节剂情况下,由配置多重检测器的凝胶渗透色谱仪获得的Mw明显大于由单一RI检测器获得的Mw,与PM通过GPC测试得到的数据一致,而在添加极性调节剂情况下,由配置多重检测器的凝胶渗透色谱仪测试得到的Mw均小于配置单一RI检测器获得的Mw,同时其dn/dc也相应提高,结合光散射机理,推测产生此现象的原因是在添加极性调节剂条件下,其微观结构改变导致dn/dc变化。
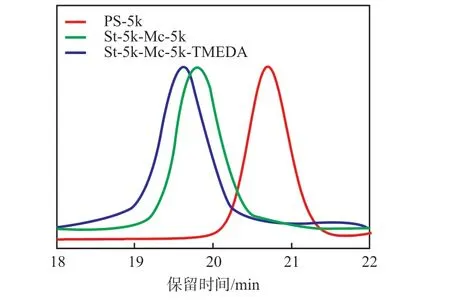
图4 添加调节剂前后SM的GPCFig.4 GPC curves of SM before and after adding modifier
2.2 PM与3-巯基丙酸甲酯的“巯基-双键”点击反应
为了进一步探究PM中不同类型双键在“巯基-双键”点击反应中的活性,将3-巯基丙酸甲酯与PM反应,在365 nm紫外光辐照下,分别以DMPA和BP为光引发剂,从图5和表3可以看出:DMPA作为光引发剂时,PM与3-巯基丙酸甲酯的“巯基-双键”点击反应得到的聚合物形成了明显的凝胶状,说明发生了交联,提高3-巯基丙酸甲酯的比例,交联现象也无法改善,只有降低反应体系中DMPA的用量,如双键与DMPA摩尔比为1.00∶0.02时,凝胶才消失。从表3还可以看出:3-巯基丙酸甲酯接枝到PM骨架中双键的接枝率为20%~30%,当延长反应时间时,聚合物的Mw/Mn明显变宽,反应位点无法控制。当BP作为光引发剂时,交联现象得到明显改善,体系中不产生凝胶。但是当PM中的双键与3-巯基丙酸甲酯的投料比例减小时,通过延长反应时间,所得到的聚合物Mw/Mn均较宽。

图5 DMPA催化条件下不同投料比及反应时间所产生的凝胶Fig.5 Gel produced by different feed ratios and reaction times under DMPA catalysis

表3 PM与3-巯基丙酸甲酯点击反应的GPC与1H-NMR数据Tab.3 GPC and 1H-NMR data of click reaction between PM and methyl 3-mercaptopropionate
从图6可以看出:当双键、3-巯基丙酸甲酯、BP的摩尔比为1.00∶10.00∶1.00,反应时间为48 h时,与接枝前相比,接枝后聚合物流出曲线的保留时间明显变小,说明3-巯基丙酸甲酯成功接枝到了PM的骨架中,并通过1H-NMR计算,得到接枝率为99%。当缩短反应时间为24 h时,3-巯基丙酸甲酯接枝到PM中双键的接枝率由99%降低到75%(见表3)。从图6还看出:δ为3.4~3.8归属于3-巯基丙酸甲酯与氧相连的甲基特征峰,4.6~5.2归属于PM骨架中双键的特征峰,根据峰位归属和积分面积计算,δ为4.6~5.2的特征峰强度消失或减弱,一方面证实了3-巯基丙酸甲酯与PM中的双键发生了“巯基-双键”点击反应,另一方面说明通过改变反应时间从48 h到24 h,接枝率发生了改变,由99%降低为75%,实现了对该“巯基-双键”点击反应的控制。
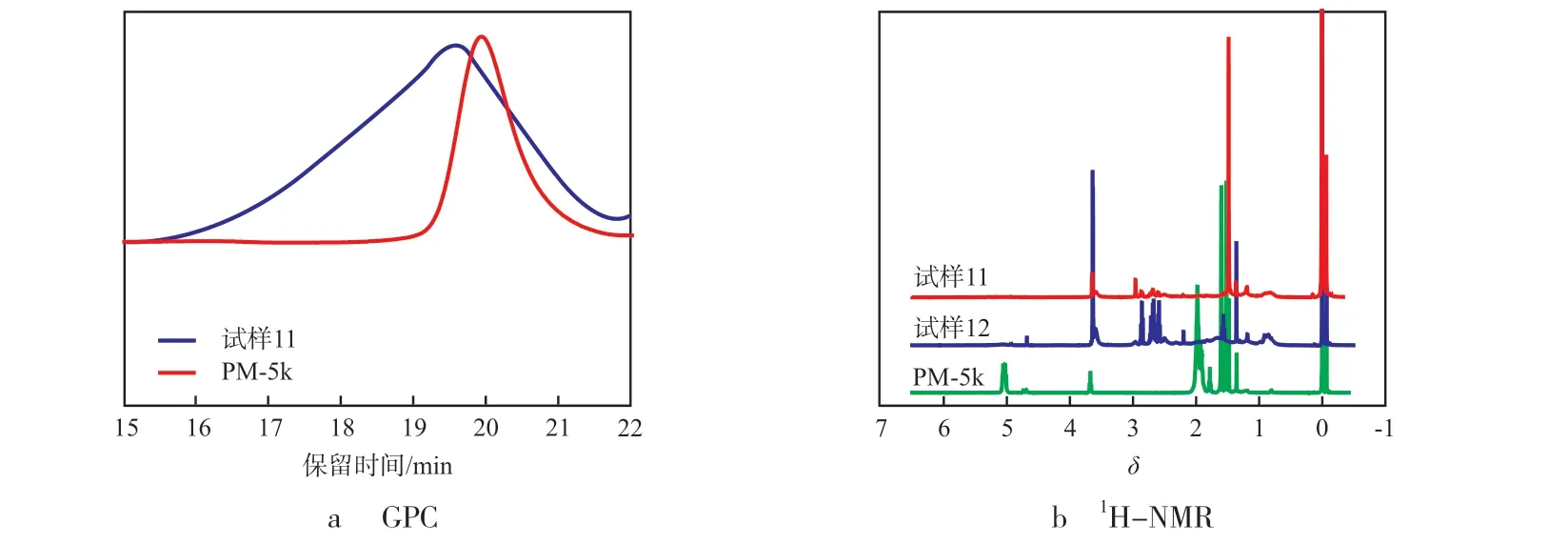
图6 接枝前后PM的GPC与1H-NMRFig.6 GPC and 1H-NMR curves of PM before and after grafting
巯基与双键的反应机理为:在紫外光辐照下,引发剂产生自由基,引发含巯基的化合物(如3-巯基丙酸甲酯)产生自由基,与此同时,双键产生自由基,因此,巯基化合物自由基与双键自由基相互进攻反应终止。当巯基化合物形成的自由基浓度较低时,双键自由基进攻双键产生连锁反应,形成分子内自交联或分子间交联;当巯基化合物浓度较高时,可以有效抑制双键自由基的交联。本工作中,DMPA作为光引发剂时,DMPA形成的自由基活性较高,当DMPA浓度较高时,快速引发高浓度的双键自由基,因此,双键自由基发生了交联形成了凝胶;当DMPA与巯基化合物浓度较低时,形成少量的双键自由基,有效地抑制了交联的发生。试样4与试样5在GPC中显示了相对窄的Mw/Mn(见表3),证实了PM骨架少量的双键发生了“巯基-双键”点击反应。相比之下,当BP作为光引发剂且浓度相对较高时,PM均未发生交联而形成凝胶,说明BP自由基的活性较低,未能引发高浓度双键自由基的形成,有效地抑制了双键间的交联,因此通过改变反应时间,可以实现接枝率的控制。巯基与双键的反应机理见图7。
3 结论
a)采用阴离子聚合方法,以sec-BuLi为引发剂,成功合成了PM,SM,SMS。
b)在添加TMEDA[n(sec-BuLi)∶n(TMEDA)=1∶1]的条件下,采用一步投料法获得了以1,2-聚合为主的SM。
c)以DMPA为引发剂,高浓度引发剂导致交联,低浓度的DMPA和巯基化合物有效抑制交联,但接枝率低(20%~30%),通过延长反应时间,得到的聚合物Mw/Mn宽。
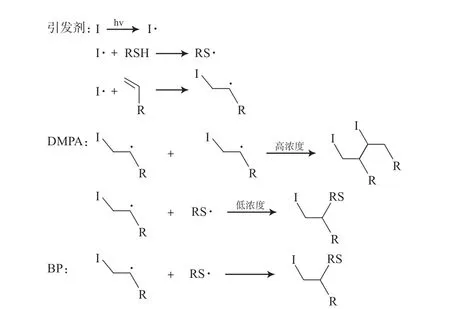
图7 不同光引发剂催化巯基点击反应原理Fig.7 Mechanism of thiol-ene click reaction catalyzed by different photocatalysts
d)以低活性BP为光引发剂,不发生交联现象,提高巯基化合物比例可有效控制点击反应。在双键、巯基、引发剂摩尔比为1.00∶10.00∶1.00的条件下,反应48 h可以实现99%的接枝率,且聚合物Mw/Mn较窄。
e)采用阴离子聚合方法合成了PM,SM,SMS,研究了其阴离子反应特性,并分析了PM的“双键-巯基”点击功能化特点,对合成绿色生物基橡胶具有重要意义。
猜你喜欢
中学化学(2022年6期)2022-07-05
油气·石油与天然气科学(2021年12期)2021-12-11
中国金属通报(2021年20期)2021-03-11
中国化妆品(2020年6期)2020-07-22
商品与质量(2019年31期)2019-11-28
科技视界(2019年25期)2019-11-19
中国新技术新产品(2019年5期)2019-05-21
丝路视野(2018年8期)2018-05-14
分析化学(2017年12期)2017-12-25
魅力中国(2017年23期)2017-10-14
