
利用CRISPR/Cas9 AAV系统构建纹状体Slc20a2基因敲除小鼠模型
2020-10-29 05:48林珉婷赖璐璐赵淼林必玮姚香平
遗传 2020年10期
林珉婷,赖璐璐,赵淼,林必玮,姚香平,3
利用CRISPR/Cas9 AAV系统构建纹状体基因敲除小鼠模型
林珉婷1,2,赖璐璐1,赵淼1,林必玮1,姚香平1,3
1. 福建医科大学附属第一医院神经内科,福州 300005 2. 福建医科大学神经科学研究院,福州 300004 3. 福建省分子神经病学重点实验室,福州 300005
原发性家族性脑钙化症(primary familial brain calcification, PFBC)是慢性进展性的神经系统遗传病,临床症状主要包括运动障碍、认知障碍及精神障碍等,其致病机制尚未完全明确。研究表明是该病最主要的致病基因。由于基因全身性敲除小鼠模型会导致胎儿生长受限,为更好地研究PFBC发病机制,本研究应用CRISPR/Cas9技术构建了纹状体基因条件性敲除小鼠模型。首先,针对基因编码区,设计3条靶向exon3的sgRNA (single guide RNA),通过构建质粒、转染细胞、Surveyor assay等实验验证sgRNA的活性。其次,选取活性较高的sgRNA重组包装AAV-Cre病毒,应用立体定位将AAV病毒定点注射于小鼠纹状体。体外实验结果表明设计的3条sgRNA均能够有效地介导Cas9切割靶DNA。细胞免疫荧光实验结果证实AAV-Cre病毒具有Cre重组酶活性。最后,通过小鼠脑部组织免疫组化、TA-克隆、高通量测序及Western blot方法检测基因敲除效率,发现实验组小鼠纹状体组织表达明显降低。本研究成功设计了3条能够敲除的功能sgRNA,并应用CRISPR/Cas9技术成功构建了纹状体基因条件性敲除小鼠,为研究PFBC的发病机制提供了有效的动物模型。
CRISPR/Cas9;;sgRNA;基因敲除;小鼠模型
原发性家族性脑钙化症(primary familial brain calcification, PFBC)是以纹状体、丘脑、小脑、皮层等部位对称性钙化为特征的神经系统遗传病[1]。临床主要表现为运动障碍、精神症状、认知障碍、癫痫、头痛等症状。目前PFBC发病机制尚不明确,且具有高度遗传异质性[2]。迄今为止,已明确有6个PFBC致病基因:和[3~8]。其中,2基因被认为是PFBC最常见的致病基因,该基因编码III型钠–磷协同转运体2 (PiT2),突变后严重影响磷的摄取功能,引起细胞外磷酸根离子聚集,进而导致钙磷沉积[9,10]。虽然已克隆多个PFBC致病基因,但该病仍缺乏有效的治疗手段,具体的发病机制及治疗方法仍需进一步探讨。
动物模型是模拟疾病表型、研究疾病机制及探索治疗手段的良好工具。为管家基因,在体内广泛表达。基因纯合敲除小鼠表现为胚胎生长受限,且部分新生小鼠会在断奶前死亡[11,12]。PFBC患者的病理性钙化可以累及脑区各个位置,但是纹状体是最为常见的受累部位。因此,构建纹状体基因敲除小鼠模型,不但可以解决基因全身敲除小鼠胚胎生长受限的困境,而且能够特异性地研究纹状体区域脑钙化具体机制。
CRISPR/Cas9系统由于具备效率高、设计简单等优势,是目前最为广泛运用的基因编辑技术。CRISPR/Cas9系统包含两大组分:一是行使剪切功能的Cas9蛋白;另一个是特异性引导Cas9蛋白至靶DNA序列的sgRNA(single guide RNA)。由于Cas9蛋白分子量较大,且血脑屏障的存在使得Cas9蛋白病毒载体在神经系统中不易于表达。LSL-Cas9-EGFP小鼠是基因、基因敲入小鼠,可以条件性表达Cas9蛋白和EGFP蛋白[13]。通过向小鼠脑区或者核团注射sgRNA和Cre,可以激活LSL-Cas9- EGFP小鼠表达Cas9蛋白,从而实现定点目的基因的敲除。本研究运用CRISPR/Cas9技术,基于LSL-Cas9-EGFP小鼠,进行纹状体基因条件性敲除,为PFBC的发病机制及药物研究提供良好的动物模型。
1 材料与方法
1.1 材料
实验中所用小鼠为条件性表达Cas9及EGFP蛋白小鼠(LSL-Cas9-EGFP小鼠,B6;129-Gt(ROSA) 26Sortm1(CAG-Loxp-Stop-LoxP-Cas9, -EGFP)Fezh/J),由美国The Jackson Laboratory实验室引进。AAV病毒表达载体(60229, AAV:ITR-U6-sgRNA-backbone- pCBh-Cre-WPRE-hGHpA-ITR)购于美国Addgene公司,含Cre位点以及两个I酶切位点。Reporter质粒(20299, pAAV-EF1a-double floxed-mCherry-WPRE- HGHpA)购于美国Addgene公司,其中两个flox序列是反向插入,可条件性表达红色荧光蛋白。实验室常规保存质粒px458(Cas9/sgRNA/EGFP共同表达质粒)含I酶切位点。引物由生工生物工程(上海)股份有限公司合成。T4 DNA连接酶、I、I等核酸内切酶购于美国NEB公司,细胞株为小鼠神经瘤母细胞N2A细胞、293T细胞。
1.2 sgRNA设计和寡核苷酸链合成
根据CRISPR/Cas9系统sgRNA设计原则,使用Zhang lab网站(http://crispr.mit.edu)对鼠源基因(Gene ID: 20516)第3外显子(290~429 bp)设计靶点,选择敲除效率分数最高的3个sgRNA (sgRNA1、sgRNA2、sgRNA3),具体信息见表1。sgRNA序列若不以G碱基开头,可添加1个G碱基以便U6启动子转录。在合成sgRNA时需在序列里添加5ʹ-cacc-3ʹ和5ʹ-aaac-3ʹ (表1),使其退火后形成可与I酶切位点互补的粘性末端。根据靶位点设计扩增引物,分别为E3F:5ʹ-TGACTCATCATTGGCACAGG-3ʹ和E3R:5ʹ-TAAGGCTCTTCTTCACGTGG-3ʹ,扩增长度为650 bp。sgRNA1、sgRNA2和sgRNA3切割后产物大小分别约为297 bp和353 bp、288 bp和362 bp、248 bp和402 bp。
1.3 构建Slc20a2基因px458-sgRNA打靶载体
实验所用质粒载体为px458,使用I限制性内切酶酶切,37℃水浴1 h,按照胶回收试剂盒说明书回收约9300 bp长度的DNA片段。将合成的sgRNA的上游(F)和下游(R)引物用退火缓冲液稀释至9 mmol/L,用PCR仪退火,程序为95℃ 5 min,72℃ 10 min,37℃ 20 min,即退火为双链DNA,用于后续连接反应。退火后的sgRNA寡核苷酸双链与回收的px458载体片段在T4 DNA连接酶作用下16℃过夜连接,连接产物转化至DH5α感受态,挑取单克隆菌落并提取质粒px458-sgRNA1、px458- sgRNA2和px458-sgRNA3,通过一代测序验证将sgRNA序列克隆到px458质粒中。

表1 sgRNA序列
合成sgRNA时需在序列里添加5ʹ-cacc-3ʹ和5ʹ-aaac-3ʹ,以与I酶切位点的粘性末端互补。
1.4 sgRNA活性检测
将4 μg质粒px458-sgRNA用脂质体lipo3000 (L3000-015,美国Invitrogen公司)转染至汇合度达到60%~80%的N2A细胞,对照为转染px458空载质粒(空白对照),混匀,置于5% CO2、37℃孵箱中培养。转染72 h后提取细胞DNA。弃除细胞培养基,加1 mL PBS将细胞吹散,收于1.5 mL EP管中,12,000 r/min 离心1 min,去上清,留沉淀。用血液/细胞/组织基因组DNA提取试剂盒(北京天根公司)提取基因组DNA。然后吸取2 μL DNA作为模板,引物为E3F和E3R,用KOD酶(TOYOBO公司,日本)体系进行PCR扩增。扩增程序:95℃ 5 min;94℃ 30 s,60℃ 30 s,68℃ 30 s,35个循环;68℃ 5 min。用纯化试剂盒纯化PCR产物,然后进行变性、复性,其程序为95℃ 5 min,在95℃降至85℃期间,以每秒2℃退温,在85℃降至25℃期间,以每秒0.1℃退温,最后产物于4℃放置10 min。按照Surveyor突变检测试剂盒(Transgenomic公司,美国),分别加入1 μL Surveyor Nuclease S、1 μL Surveyor Enhancer S、0.15 mol/L MgCl22 μL和20 μL DNA,混匀后42℃放置1 h,加入1/10体积的终止液混匀后,进行聚丙烯酰胺凝胶电泳分离。根据编辑效率公式fcut = (b+c)/(a+b+c)和indel(%)=100×(1−(1−fcut))进行计算来评估CRISPR/Cas9的编辑效率。
1.5 构建AAV-sgRNA-Cre载体及其Cre重组酶活性验证
AAV-Cre病毒表达载体(60229,美国Addgene公司)[13],使用I限制性内切酶酶切,37℃水浴1 h,按照胶回收试剂盒说明书回收约6400 bp长度的DNA片段。将筛选出活性最高的sgRNA退火后(方法见1.3)与酶切回收的AAV病毒表达载体片段连接,转化至DH5α感受态,挑取单克隆菌落并提取质粒进行测序,验证sgRNA序列重组到AAV-Cre载体上。将重组后的AAV-Cre质粒和红色荧光蛋白报告基因质粒(AAV-double-floxed-mcherry)共转检测Cre重组酶活性。实验分成5组,分别为转染Cre-GFP质粒(阳性对照)、转染红色荧光蛋白报告基因质粒(报告质粒)、转染AAV-Cre质粒(实验组)、共转阳性对照和报告质粒以及共转实验组和报告质粒。转染前24 h,消化293T细胞铺板24孔板,每孔接种细胞1×105个。转染质粒总量为1 μg,转染试剂为Lipofectin 3000。转染后6 h进行细胞换液,弃去培养盘中原有培养基,加入0.5 mL完全培养基。转染48 h后观察荧光,进行拍照。将重组后的AAV-Cre由上海泰廷生物科技有限公司进行病毒包装、纯化和浓缩。
1.6 立体定位法注射AAV-sgRNA-Cre病毒
LSL-Cas9-EGFP成年鼠随机分为GFP病毒注射组(CON组)和AAV-Cre病毒注射组(KO组)。用戊巴比妥钠60~80 mg/kg腹腔注射麻醉成年小鼠。将小鼠两侧外耳道及牙齿固定在脑立体定向仪(Stoelting公司,美国),常规备皮消毒后进行头部剪毛,剪去表层皮肤暴露前囟并标记前囟位置。根据坐标点调整好钻孔位置,确立右侧纹状体坐标:前囟后0.46 mm,中线侧旁开2.00 mm,硬膜下4.00 mm。调整游标卡尺对应其坐标点,用牙科钻孔器钻孔。用微量注射器(上海高鸽工贸有限公司)进针,缓慢注射。注射速度0.2 μL/min,注射病毒量0.5~2.0 μL。注射完毕停10 min后缓慢退针、缝皮、消毒、解除固定。给予光照为小鼠保暖,等待小鼠苏醒。注射过程及结束后均需要观察动物的反应及有无死亡。注射后1月、2月时分别处死小鼠,进行全脑冰冻切片。注射6月后处死小鼠,提取注射部位的纹状体组织,留取标本。
1.7 免疫组化验证
分别在注射后的每个时间点取脑组织。用戊巴比妥钠60~80 mg/kg腹腔注射麻醉小鼠,固定于平板上。依次打开腹腔、胸腔、暴露心脏,用眼科剪剪破右心耳,用注射针尖刺入左心室。先用50 mL生理盐水灌注,直至肝脏变白,接着用50 mL的4%多聚甲醛进行灌注。待鼠尾巴及身体完全僵硬后,取出小鼠的大脑组织。将取出的大脑组织置于4%多聚甲醛固定24 h,接着用30%的蔗糖脱水,待大脑沉入容器底部行冰冻切片。用冰冻切片机(Leica公司,德国)进行冠状位切片,切片厚度为40 μL。用GFP、Cre抗体进行脑片免疫组化。用封闭液配一抗GFP (1:2000,美国Invitrogen公司)和Cre (1:2000,美国Milipore公司),4℃孵育一抗过夜。次日用1×PBS轻轻洗涤3次,各5 min。用封闭液配相应的二抗,于室温下避光120 min,1×PBS轻轻洗涤3次。用DAPI (1:5000,上海碧云天生物技术有限公司)覆盖细胞表面,1×PBS洗涤3次。在载玻片上滴一滴封片剂,将爬片的细胞面扣在封片剂上。于共聚焦显微镜(Zeiss公司,德国)下观察,并拍照。
1.8 流式分选富集Slc20a2基因点突变细胞
取AAV-sgRNA-Cre病毒注射6个月的LSL-Cas9- EGFP小鼠,用戊巴比妥钠麻醉处死后断颈,取出右侧纹状体。充分剪碎后,加入胰酶消化并吹打数次。置于37℃ 10 min。待充分消化后,加入含FBS培养基终止消化,离心1000 r/min 5min。用细胞培养基重悬细胞,于流式细胞仪(Beckman公司,美国)筛选带GFP细胞。提取筛选后细胞的基因组,由于组织量较少,需用KAPA快速DNA提取试剂盒(KAPA公司,上海)进行DNA提取。Nested-PCR扩增获取基因,第一次扩增引物为上述的E3F、E3R。第二次扩增引物为F:5ʹ-GTACATTGTCGATGCTCTCC-3ʹ;R:5ʹ-GAAAGGGTGGCGTTAAACCTG-3ʹ。第二次扩增后的产物经2%琼脂糖胶电泳,鉴定单一特异性条带后送公司测序评估基因编辑效果。测序峰图上在sgRNA识别的NGG序列附近如果出现多层套叠峰时,表明该注射病毒的脑区发生基因编辑。将上述检测到发生突变的PCR产物连接pMD-18T载体中,转化涂板。培养过夜后,挑单克隆(r1~r7等)进行测序鉴定获得具体的基因编辑后的突变类型。
1.9 高通量扩增子测序
取AAV-sgRNA-Cre病毒注射后6个月的LSL- Cas9-EGFP小鼠,用戊巴比妥钠麻醉处死后断颈,取出脑组织。分为左侧大脑半球(自身对照组)和右侧大脑半球(实验组)两个部分。待充分剪碎后,提取组织DNA,扩增获取基因。第一次扩增引物为F1:5ʹ-GCCAGGTGGCTAAGACACTG-3ʹ和R1:5ʹ- CCTTGCTCCGTTGCCTATAC-3ʹ。为进行后续的二代测序,需进行二次扩增,在引物5ʹ端加接头。左半球脑组织的第二次扩增引物为F2:5ʹ-GAATTCTCAATGCCGGATCCGCCAGGTGGCTAAGACACTG-3ʹ和R2:5ʹ-GAATTCTGGCGATAGGATCCCCTTGCTCCGTTGCCTATAC-3ʹ,右半球脑组织的第二次扩增引物为F3:5ʹ-GAATTCCAAGCACCGGATCCGCCAGGTGGCTAAGACACTG-3ʹ和R3:5ʹ-GAATTCTTTCGACGGGATCCCCTTGCTCCGTTGCCTATAC-3ʹ。扩增后的产物经2%琼脂糖胶鉴定单一特异性条带后送金唯智公司进行高通量扩增子测序。使用MiSeq平台测序DNA文库混合后,按Illumina MiSeq (Illumina公司,美国)仪器使用说明书进行2×250/ 2×250 bp 双端测序(PE),由MiSeq自带的MiSeq Control Software (HCS)+OLB+GAPipeline-1.6读取序列信息。使用fastp(v0.19.6)软件的默认参数对双端250 bp的测序原始数据进行去除测序接头和过滤低质量reads的处理。随后,根据样本的barcode信息对上一步得到的FASTQ文件(clean reads)进行拆分。双端reads的拼接、比对以及比对后结果的统计使用CRISPResso2软件的默认参数完成。
1.10 Western blot检测
戊巴比妥钠麻醉分别处死实验组小鼠(KO组)、对照组小鼠(CON组)、未注射病毒的空白组小鼠(WT组)。取出右侧纹状体,充分剪碎后,加入800 μL LBA150裂解缓冲液(30 mmol/L Tris-HCl pH 7.4、150 mmol/L NaCl、0.5% NP40、10% glycerinum、1 mmol/L EDTA、1 mmol/L EGTA、1 mmol/L PMSF、1 mmol/L Cocktail、1 mmol/L NaF、1 mmol/L Na2NO3)。继续研磨至无明显组织形态的溶液状,于冰上裂解30 min。转至新的EP管中,4℃ 12,000 r/min离心30 min。取上清做好标记部分保存于–80℃。取适量用于蛋白浓度的测定,按照BCA法(上海碧云天生物技术有限公司)蛋白浓度。加入5×SDS Loading Buffer,100℃水浴10 min,14,000 r/min离心5 min ,在12%浓度分离胶的SDS-PAGE胶中电泳。电泳结束后小心剥下凝胶,采用湿转法将蛋白转移到PVDF膜上,室温封闭1 h。4℃孵育一抗(Anti-PiT2,英国Abcam公司)过夜。孵育完毕用TBST洗膜3次,随后室温孵育二抗1 h,孵育完毕用TBST洗膜3次,采用ECL法在化学发光自显影仪检测蛋白条带并拍照。
2 结果与分析
2.1 CRISPR/Cas9打靶载体的活性检测,筛选高效sgRNA
根据CRISPR/Cas9靶点设计原则,sgRNA序列选在基因的E3外显子(图1 A),将选择后的3条sgRNA序列重组至px458质粒上。经Sanger测序验证,成功构建基因的Cas9打靶质粒(图1,B~D)。将构建好的CRISPR/Cas9打靶质粒px458- sgRNA1、px458-sgRNA2和px458-sgRNA3及阴性对照质粒px458转染N2A细胞,按照 Surveyor突变检测试剂盒说明书进行检测,发现均出现两条及以上的新条带(图1E)。sgRNA1酶切后的新片段长度预测约为297 bp和353 bp,sgRNA2约为288 bp和362 bp,sgRNA3约为248 bp和402 bp。根据文献[13]的编辑效率公式,对条带灰度值进行计算,结果表明3条sgRNA均靶向敲除成功,其切割效率分别为52.2%、42.3%和46.8%,其中sgRNA1活性最强,可用于后续实验。
2.2 体外鉴定病毒载体Cre重组酶活性
将筛选出来的sgRNA序列重组至AAV-Cre病毒载体上,通过与红色荧光蛋白报告基因质粒(AAV- double-floxed-mCherry)共同转染293T细胞,检测其Cre重组酶的活性。结果发现分别单转阳性对照(Cre-GFP)、报告质粒和实验组(AAV-Cre)时均未观察到红色荧光,当实验组和阳性对照分别与报告质粒共同转染时均可观察到红色荧光,表明实验组病毒载体具备Cre重组酶活性(图2)。

图1 体外打靶效率检测
A:打靶质粒靶点示意图(靶点序列为红色标记,绿色标记的是PAM序列);B~D:重组测序图(靶点sgRNA序列为红色标记,绿色标记的是px458质粒上的sgRNA scaffold序列的部分);E:打靶效率检测(a为PCR扩增的主带,大约650 bp;b、c分别是主带切割形成的预期大小的条带)。M为DNA Maker;Mock为空白对照。
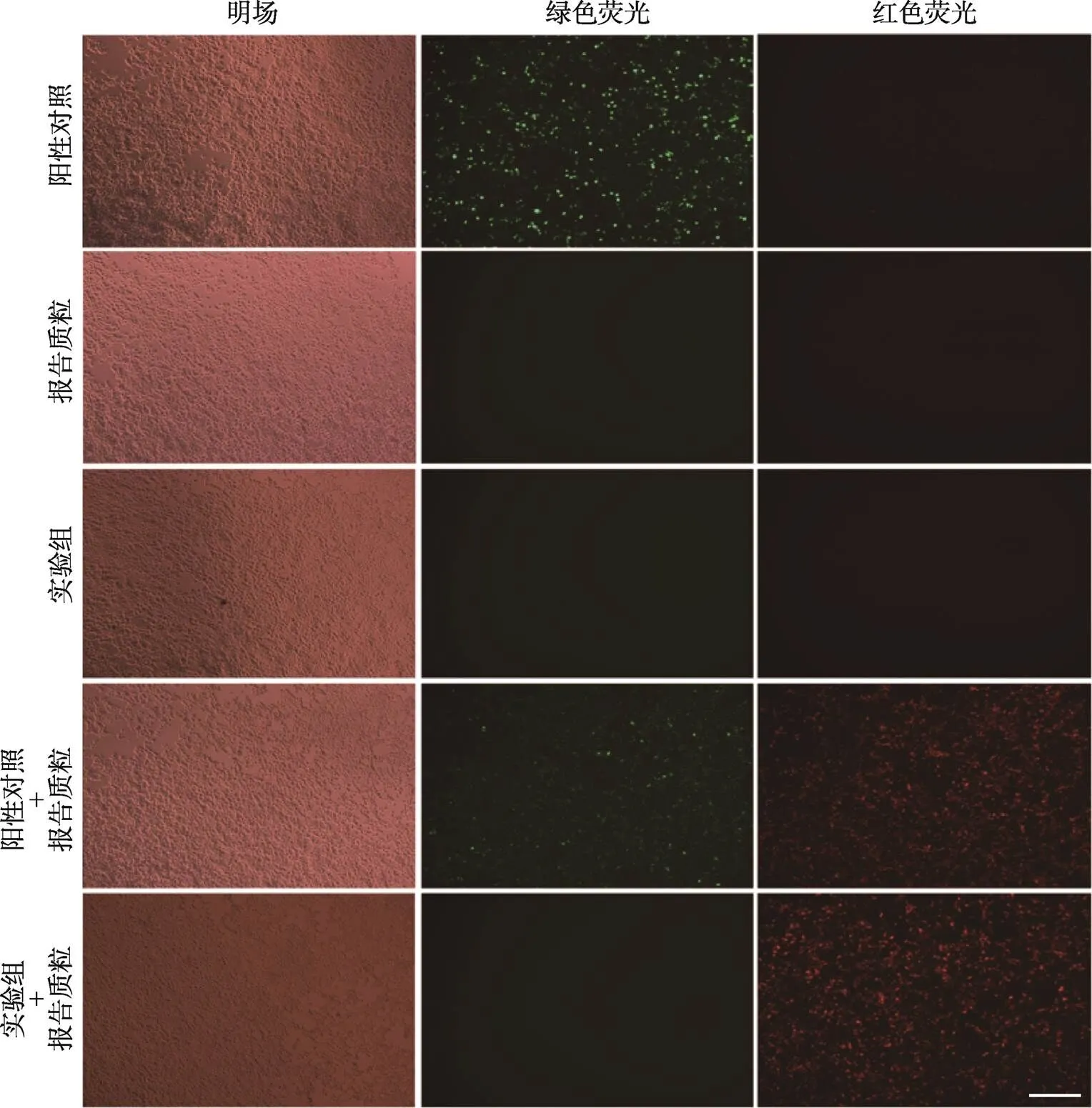
图2 Cre重组酶活性体外实验验证
实验组与报告质粒共转染时可观察到红色荧光,表明实验组AAV-Cre病毒载体具备Cre重组酶活性。标尺=100 μm。
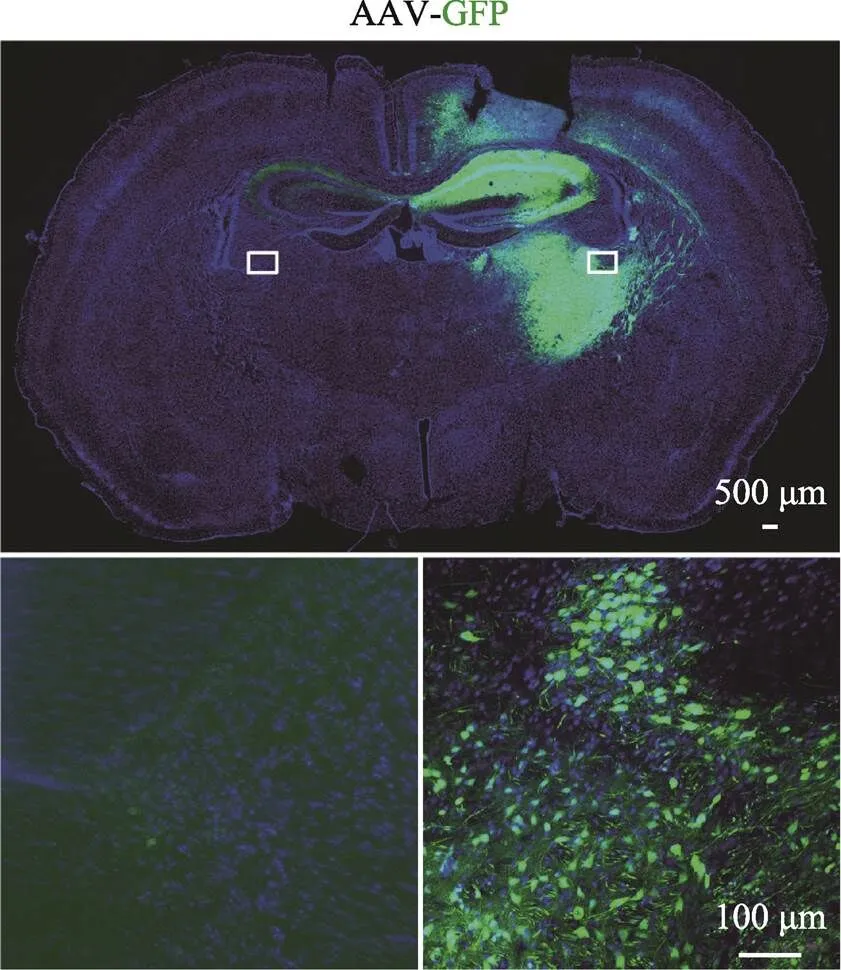
图3 对照组脑组织免疫荧光图
上图为对照组小鼠在注射AAV-GFP病毒2周后的脑组织免疫荧光图,注射部位出现绿色荧光。标尺=500 μm;下图为上图白色小方框的放大示意图,标尺=100 μm。
2.3 纹状体Slc20a2基因敲除小鼠模型构建
取2~3月的LSL-Cas9-EGFP小鼠进行右侧纹状体病毒注射。对照组注射AAV-GFP病毒,实验组注射AAV-Cre病毒。在病毒注射后2周、1个月、2个月分别进行免疫组化检测,发现AAV-GFP对照组右侧纹状体部位出现GFP荧光,弥散分布于纹状体大部分,蔓延至脑室(图3)。AAV-Cre实验组可以观察到注射部位出现表达Cre重组酶的红色荧光和表达Cas9-EGFP蛋白的绿色荧光(图4)。在病毒注射后6个月,提取小鼠的右侧纹状体组织,进行流式分选GFP阳性细胞。通过PCR扩增基因靶点区域,经测序显示靶点区域出现套峰(图5A),说明基因被编辑。进一步进行TA克隆观察到在靶点区域出现碱基的随机插入、缺失或替换,编辑效率约为52.78% (19/36) (图5B)。通过提取AAV-Cre病毒注射6个月后的小鼠脑组织,扩增靶点区域后进行高通量扩增子测序,结果显示右侧大脑半球的基因编辑效率为5.99%,明显高于左侧大脑半球的基因编辑效率(0.36%) (图5C)。右侧大脑半球在sgRNA结合的序列附近出现较高频率的碱基插入、缺失或替换。通过提取实验组小鼠右侧纹状体组织,Western blot结果显示,实验组(KO)PiT2蛋白表达显著低于对照组(CON)与空白组(WT),约为对照组的21% (图5,D和E)。通过对AAV-Cre病毒注射6个月后的小鼠进行微型CT检查和病理切片染色,均未检测到病理性的脑钙化结节。

图4 实验组脑组织免疫荧光图
上图为实验组小鼠在注射AAV-Cre病毒2周后的脑组织免疫荧光图,注射部位出现红色荧光和绿色荧光。标尺=500 μm。下图为上图白色小方框的放大示意图。图中的箭头分别表示可观察到表达Cre重组酶的红色荧光、表达Cas9-EGFP蛋白的绿色荧光以及同时表达两者的荧光。标尺=100 μm。

图5 纹状体Slc20a2基因敲除验证
A:实验组小鼠右侧纹状体组织经GFP抗体筛选后的PCR测序图显示sgRNA序列附近出现套峰;B:实验组小鼠右侧纹状体组织经GFP抗体筛选后的TA克隆基因型显示PAM序列附近碱基的随机插入或缺失;C:实验组左右大脑半球高通量扩增子测序结果比较,右侧大脑半球有更高的基因编辑效率;D:实验组、对照组及空白组小鼠的右侧纹状体组织PiT2蛋白免疫印迹图;E:图D的结果统计图。实验组PiT2蛋白的表达显著低于对照组、空白组。图中数值给出的是平均值±标准误,=3。**:<0.01;***:<0.001;NS表示无统计学意义。
3 讨论
目前,有研究报道关于基因全身性敲除小鼠模型,表现颅脑钙化的同时多合并其他的症状,如胚胎生长受限、视神经钙化、脑积水[12,14,15]。推测可能的机制是纯合型–/–的临床表型比杂合型+/–明显。作为致病基因为常染色体显性遗传,临床上除了一例报道复合杂合突变的患者临床症状较其父母(均携带杂合突变)严重许多外[3],其余PFBC患者均为杂合突变。基因致病的机制是由于编码的PiT2突变后降低细胞对无机磷的摄取,推测杂合突变导致单倍体剂量不足而致病。基因作为管家基因,不仅参与大脑的磷代谢过程,在身体其他部位的磷代谢中也起重要作用[16~18]。所以由于基因全身性敲除小鼠模型的局限性,组织特异性的基因敲除小鼠成为研究PFBC的关键。
本研究通过CRISPR/Cas9技术构建纹状体基因敲除小鼠,不仅可以解决全基因敲除小鼠致死或发育不良的困境,而且能够特异性地研究纹状体钙化的具体机制。TA克隆的基因型分析发现Cas9蛋白对靶点进行切割后,细胞对其修复的过程中导致碱基随机的插入或缺失,导致基因突变,从而模拟临床PFBC的发病过程。通过对病毒注射部位行流式分选GFP阳性细胞,由于可以富集经基因编辑的细胞,编辑效率明显高于在体的编辑效率。但本研究中的小鼠脑区基因编辑效率很可能被低估,原因在于病毒的注射部位局限于小鼠的右侧纹状体组织,而进行高通量测序时采用的样品为一侧大脑半球。从免疫荧光(图3,图4)结果可以看出病毒高度集中在注射部位,标本的选择是导致基因编辑效率被低估的关键。本研究在体基因编辑效率仍有待于提高,造成这种情况的原因主要是立体定位注射技术操作难度大、AAV病毒浓度的调整、AAV病毒Cre重组酶的活性等都是非常重要的因素。
本研究结合Cre-LoxP系统和CRISPR/Cas9技术,采用立体定位的方法将AAV-Cre病毒注射至LSL- Cas9-EGFP小鼠的右侧纹状体。当病毒的Cre重组酶表达后切除CAG启动子下游loxP位点之间的stop序列,激活Cas9-EGFP的表达。表达后的Cas9蛋白进而在sgRNA的引导下产生基因编辑作用。而常见的Cre-LoxP条件性敲除的Cre重组酶往往被置于某特定基因启动子的下游,目的基因的两端分别含有一个loxP位点,从而实现较高的组织和细胞特异性。传统的条件性敲除往往需要较长的实验周期、操作复杂[19],而本研究的模型构建方法设计较为巧妙,更为简便、有效。CRISPR/Cas系统由于脱靶率高在一定程度上限制了其应用[20]。结合AAV-Cre病毒的定点注射在实现组织特异性敲除的同时可以有效地避免非靶细胞编辑的脱靶效应。实验组的PiT2蛋白表达量比对照组明显降低,进一步证实纹状体基因被敲减。但半年后对实验组小鼠进行病理切片染色或微型CT检查,未显示出病理性的脑钙化结节,推测可能的原因是形成病理性的钙化结节需要一定的时间。有研究表明全基因敲除小鼠需要至少5个月甚至更长的时间[21],且钙化病灶往往随着年龄的增长而增大。基因敲减后是否出现与疾病相似的表型需要更长的时间,有待于后续实验进一步研究。
小鼠纹状体基因敲除的模型构建在国内外未见报道,本研究利用CRISPR/Cas9技术联合Cre-LoxP系统成功构建了基因条件性敲除小鼠,为今后探究PFBC的钙磷代谢机制及可能存在的治疗研究提供良好的模型。
[1] Nicolas G, Pottier C, Charbonnier C, Guyant-Marechal L, Le Ber I, Pariente J, Labauge P, Ayrignac X, Defebvre L, Maltete D, Martinaud O, Lefaucheur R, Guillin O, Wallon D, Chaumette B, Rondepierre P, Derache N, Fromager G, Schaeffer S, Krystkowiak P, Verny C, Jurici S, Sauvee M, Verin M, Lebouvier T, Rouaud O, Thauvin-Robinet C, Rousseau S, Rovelet-Lecrux A, Frebourg T, Campion D, Hannequin D. Phenotypic spectrum of probable and genetically-confirmed idiopathic basal ganglia calcification., 2013, 1369(11): 3395–3407.
[2] Oliveira JRM, Spiteri E, Sobrido MJ, Hopfer S, Klepper J, Voit T, Gilbert J, Wszolek ZK, Calne DB, Stoessl AJ, Hutton M, Manyam BV, Boller F, Baquero M, Geschwind DH. Genetic heterogeneity in familial idiopathic basal ganglia calcification (Fahr disease)., 2004, 63(11): 2165–2167.
[3] Wang C, Li YL, Shi L, Ren J, Patti M, Wang T, de Oliveira JRm, Sobrido MJ, Quintáns B, Baquero M, Cui XN, Zhang XY, Wang LQ, Xu HB, Wang JH, Yao J, Dai XH, Liu J, Zhang L, Ma HY, Gao Y, Ma XX, Feng SL, Liu MG, Wang QK, Forster IC, Zhang X, Liu JY. Mutations inlink familial idiopathic basal ganglia calcification with phosphate homeostasis., 2012, 44(3): 254–256.
[4] Nicolas G, Pottier C, Maltete D, Coutant S, Rovelet- Lecrux A, Legallic S, Rousseau S, Vaschalde Y, Guyant- Marechal L, Augustin J, Martinaud O, Defebvre L, Krystkowiak P, Pariente J, Clanet M, Labauge P, Ayrignac X, Lefaucheur R, Le Ber I, Frébourg T, Hannequin D, Campion D. Mutation of thegene as a cause of idiopathic basal ganglia calcification., 2013, 80(2): 181–187.
[5] Keller A, Westenberger A, Sobrido MJ, Garcia-Murias M, Domingo A, Sears RL, Lemos RR, Ordoñez-Ugalde A, Nicolas G, da Cunha JE, Rushing EJ, Hugelshofer M, Wurnig MC, Kaech A, Reimann R, Lohmann K, Dobričić V, Carracedo A, Petrović I, Miyasaki JM, Abakumova I, Mäe MA, Raschperger E, Zatz M, Zschiedrich K, Klepper J, Spiteri E, Prieto JM, Navas I, Preuss M, Dering C, Janković M, Paucar M, Svenningsson P, Saliminejad K, Khorshid HR, Novaković I, Aguzzi A, Boss A, Le Ber I, Defer G, Hannequin D, Kostić VS, Campion D, Geschwind DH, Coppola G, Betsholtz C, Klein C, Oliveira JR. Mutations in the gene encoding PDGF-B cause brain calcifications in humans and mice.,2013, 45(9): 1077–1082.
[6] Legati A, Giovannini D, Nicolas G, López-Sánchez U, Quintáns B, Oliveira JR, Sears RL, Ramos EM, Spiteri E, Sobrido MJ, Carracedo A, Castro-Fernández C, Cubizolle S, Fogel BL, Goizet C, Jen JC, Kirdlarp S, Lang AE, Miedzybrodzka Z, Mitarnun W, Paucar M, Paulson H, Pariente J, Richard AC, Salins NS, Simpson SA, Striano P, Svenningsson P, Tison F, Unni VK, Vanakker O, Wessels MW, Wetchaphanphesat S, Yang M, Boller F, Campion D, Hannequin D, Sitbon M, Geschwind DH, Battini JL, Coppola G. Mutations incause primary familial brain calcification associated with altered phosphate export., 2015, 47(6): 579–581.
[7] Yao XP, Cheng XW, Wang C, Zhao M, Guo XX, Su HZ, Lai LL, Zou XH, Chen XJ, Zhao YY, Dong EL, Lu YQ, Wu S, Li XJ, Fan GF, Yu HJ, Xu JF, Wang N, Xiong ZQ, Chen WJ. Biallelic mutations incause autosomal recessive primary familial brain calcification., 2018, 98(6): 1116–1123.e5.
[8] Cen ZD, Chen Y, Chen S, Wang H, Yang DH, Zhang HM, Wu HW, Wang LB, Tang SY, Ye J, Shen J, Wang HT, Fu F, Chen XH, Xie F, Liu P, Xu X, Cao JZ, Cai P, Pan QQ, Li JY, Yang W, Shan PF, Li YZ, Liu JY, Zhang BR, Luo W. Biallelic loss-of-function mutations incause primary familial brain calcification., 2020, 143(2): 491–502.
[9] Lemos RR, Ramos EM, Legati A, Nicolas G, Jenkinson EM, Livingston JH, Crow YJ, Campion D, Coppola G, Oliveira JRM. Update and mutational analysis of: a major cause of primary familial brain calcification.,2015, 36(5): 489–495.
[10] Yamada M, Tanaka M, Takagi M, Kobayashi S, Taguchi Y, Takashima S, Tanaka K, Touge T, Hatsuta H, Murayama S, Hayashi Y, Kaneko M, Ishiura H, Mitsui J, Atsuta N, Sobue G, Shimozawa N, Inuzuka T, Tsuji S, Hozumi I. Evaluation ofmutations that cause idiopathic basal ganglia calcification in Japan.,2014, 82(8): 705–712.
[11] Jensen N, Schrøder HD, Hejbøl EK, Füchtbauer EM, de Oliveira JR, Pedersen L. Loss of function ofassociated with familial idiopathic basal ganglia calcification in humans causes brain calcifications in mice., 2013, 51(3): 994–999.
[12] Wallingford MC, Gammill HS, Giachelli CM.deficiency results in fetal growth restriction and placental calcification associated with thickened basement membranes and novel CD13 and lamininα1 expressing cells.,2016, 16(1): 13–26.
[13] Platt RJ, Chen SD, Zhou Y, Yim MJ, Swiech L, Kempton HR, Dahlman JE, Parnas O, Eisenhaure TM, Jovanovic M, Graham DB, Jhunjhunwala S, Heidenreich M, Xavier RJ, Langer R, Anderson DG, Hacohen N, Regev A, Feng GP, Sharp PA, Zhang F. CRISPR-Cas9 knockin mice for genome editing and cancer modeling.,2014, 159(2): 440–455.
[14] Wallingford MC, Chia JJ, Leaf EM, Borgeia S, Chavkin NW, Sawangmake C, Marro K, Cox TC, Speer MY, Giachelli CM.deficiency in mice leads to elevated phosphate levels in cerbrospinal fluid and glymphatic pathway-associated arteriolar calcification, and recapitulates human idiopathic basal ganglia calcification., 2017, 27(1): 64–76.
[15] Bezerra DP, Oliveira JRM. New studies on knockout mouse for thegene show much more than brain calcifications.,2016, 59(4): 565–566.
[16] Forster I, Hernando N, Sorribas V, Werner A. Phosphate transporters in renal, gastrointestinal, and other tissues.,2011, 18(2): 63–76.
[17] Villa-Bellosta R, Ravera S, Sorribas V, Stange G, Levi M, Murer H, Biber J, Forster IC. The Na+-Pi cotransporter PiT-2 () is expressed in the apical membrane of rat renal proximal tubules and regulated by dietary Pi., 2009, 296(4): F691–699.
[18] Villa-Bellosta R, Sorribas V. Compensatory regulation of the sodium/phosphate cotransporters NaPi-IIc () and Pit-2 () during Pi deprivation and acidosis., 2010, 459(3): 499–508.
[19] Wang JP, Zhang YM. The application of Red/ET recombination to high efficient gene-targeting vector construction., 2005, 27(6): 953–958.王军平, 张友明. Red/ET重组在基因打靶载体快速构建中的应用. 遗传, 2005, 27(6): 953–958.
[20] He XB, Gu F. Genome-editing: focus on the off-target effects., 2017, 33(10): 1757–1775.何秀斌, 谷峰. 基因组编辑脱靶研究进展. 生物工程学报, 2017, 33(10): 1757–1775.
[21] Jensen N, Schrøder HD, Hejbøl EK, Thomsen JS, Brüel A, Larsen FT, Vinding MC, Orlowski D, Füchtbauer EM, Oliveira JRM, Pedersen L. Mice knocked out for the primary brain calcification-associated geneshow unimpaired prenatal survival but retarded growth and nodules in the brain that grow and calcify over time.,2018, 188(8): 1865–1881.
Construction of a striatum-specificgene knockout mice model by CRISPR/Cas9 AAV system
Minting Lin1,2, Lulu Lai1, Miao Zhao1, Biwei Lin1, Xiangping Yao1,3
Primary familial brain calcification (PFBC) is a chronic progressive neurogenetic disorder. Its clinical symptoms mainly include dyskinesia, cognitive disorder and mental impairment; and the pathogenesis remains unclear. Studies have shown thatis the most common pathogenic gene of the disease. Since thegene knockout mouse model could result in fetal growth restriction, in order to better understand the pathogenesis of PFBC, the present study used the CRISPR/Cas9 technology to construct a conditional knockout model ofgene in the striatum of mice. First, three sgRNAs (single guide RNAs) were designed to target the exon3 ofgene. The activity of the respective sgRNA was verified by constructing expression plasmids, transfecting cells and Surveyor assay. Second, the SgRNA with the highest activity was selected to generate the recombinant AAV-Cre virus, which was injected into the striatum of mice by stereotactic method.experiments showed that the three sgRNAs could effectively mediate Cas9 cleavage of the respective target DNA. The activity of Cre recombinase of the AAV-Cre was confirmed by immunofluorescence assay. Immunohistochemistry, TA clone, high-throughput sequencing and Western blot were used to detect and evaluate the efficiency ofgene knockout. The results showed that theexpression in the striatum of mice in the experimental group decreased significantly.In this study, three sgRNAs capable of knockout ofwere successfully designed, and the conditional knockout of thegene in the striatum of mouse was successfully established by the CRISPR/Cas9 technology, thereby providing an effective animal model for studying the pathogenesis of PFBC.
CRISPR/Cas9;; sgRNA; gene knockout; mice model
2020-05-18;
2020-08-18
国家自然科学基金青年科学基金项目(编号:81801129)和福建医科大学启航基金项目(编号:2017XQ1071, 2018QH2035)资助[Supported by the National Natural Science Foundation of China (No.81801129), and Startup Fund for Scientific Research of Fujian Medical University (Nos.2017XQ1071, 2018QH2035)]
林珉婷,本科,助理研究员,研究方向:神经系统遗传病。E-mail: mintinglin@fjmu.edu.cn
姚香平,博士,主治医师,硕士生导师,研究方向:神经系统遗传病分子机制。E-mail: 119373522@qq.com
10.16288/j.yczz.20-138
2020/10/13 16:04:24
URI: https://kns.cnki.net/kcms/detail/11.1913.R.20201012.1713.002.html
(责任编委: 谷峰)
猜你喜欢
中国体育科技(2022年1期)2022-03-10
江西农业学报(2021年4期)2021-04-20
中国生殖健康(2020年4期)2021-01-18
食品科学(2020年4期)2020-03-11
中国现代中药(2019年5期)2019-07-03
科海故事博览·下旬刊(2019年6期)2019-04-16
中国生殖健康(2018年4期)2018-11-06
中国运动医学杂志(2016年3期)2016-07-10
中国组织化学与细胞化学杂志(2016年4期)2016-02-27
中国当代医药(2015年9期)2015-03-01
