
系统性红斑狼疮并发IgG4相关性疾病可能一例
2020-11-06 04:27赵丽伶张倩茹黄彦弘
中华临床免疫和变态反应杂志 2020年4期
赵丽伶,张倩茹,黄彦弘
IgG4相关性疾病(IgG4-RD)近年来逐渐被认为是一组独立的疾病,可表现为唾液腺、胰腺、肾脏等几乎任何脏器的纤维炎性病灶[1]。系统性红斑狼疮(SLE)同样也是一组多器官受累的自身免疫性疾病。虽然两组疾病之间存在一定的重叠,但SLE并发IgG4-RD的病例较为少见,该组患者的临床特征、治疗反应、预后等尚缺乏相关研究数据。通过检索PubMed、Embase、万方数据库和维普数据库,共发现5例SLE并发IgG4-RD可能的报道。现报道一例以颈部肿物、脱发为主要临床表现,血清IgG4显著升高、白细胞下降、低补体血症、多种自身抗体阳性,影像学提示多器官肿大,病理提示弥漫淋巴细胞浸润,最终诊断为SLE并发IgG4-RD可能的病例。
1 病例摘要
患者女,52岁,主因“颈部肿物、脱发3年,吞咽困难1.5年,加重20 d”于2019年11月11日收入北京清华长庚医院。患者3年前无明显诱因出现颈部肿物伴脱发、乏力,颈部超声示左颈部及左颌下异常淋巴结,双侧甲状腺增大、弥漫性病变。外院行活检示甲状腺病理:嗜酸性滤泡上皮增生结节,伴有淋巴细胞增生;淋巴结病理:慢性炎性淋巴组织反应性增生,考虑“淋巴结炎”,抗感染效果不佳。2年前自觉左颌下肿大,未在意。1.5年前出现声嘶伴吞咽困难,北京清华长庚医院外科查血常规、肝肾功能大致正常,球蛋白显著升高(40.2 g/L),甲状腺功能正常,促甲状腺激素(TSH)正常,抗甲状腺球蛋白抗体(Tg-Ab)>500 kU/L,颈胸部计算机断层扫描(CT)示左侧颌下腺区见团块状影,右侧下颌下腺未见显示,双侧颈血管鞘周围及锁骨上窝见多发结节影,甲状腺弥漫性肿大,密度减低,纵隔、双侧腋窝、心膈角区多发淋巴结轻度增大,甲状腺弥漫性肿大,行“右叶甲状腺及峡部切除术+左颈血管旁淋巴结切除术+左下颌下腺探查术”。甲状腺病理:桥本甲状腺炎;淋巴结病理:反应性增生。下颌下腺病理:间质纤维组织增生,多量淋巴细胞、浆细胞浸润(图1)。术后声嘶缓解,长期口服左甲状腺素钠片100 μg每日1次。20 d前患者左颌下肿物再次无痛性增大,否认发热、皮疹、关节痛、口眼干、口腔溃疡、光过敏消瘦等,为明确诊断收入本科。既往史:高血压2年,平素血压控制可;15个月前无明显诱因左大腿局部无痛性隆起硬结,表面无红肿、破溃。个人史、婚育史、家族史无特殊。体格检查:左侧颌下肿物,大小约3 cm×4 cm, 质韧,无压痛;双侧颈部、腋窝、腹股沟多发淋巴结肿大,最大约2 cm×1 cm,质韧,无压痛。双肺呼吸音清,未闻及干湿啰音,心律齐,各瓣膜区未闻及病理性杂音,腹软,无压痛、反跳痛,双肾区叩击痛阴性,肠鸣音正常。

图 1 左侧下颌下腺组织病理
诊治经过:入院后查血常规,白细胞 3.26(3.5~9.5)×109/L,血红蛋白112(115~150)g/L,血小板231(125~350)×109/L,嗜酸性粒细胞比例及计数正常。尿常规:白细胞 500 cell/μL,镜检白细胞 10~15/高倍视野(HPF),亚硝酸盐(-),蛋白trace(±),潜血(-),24 h尿蛋白160.80 mg/24 h,尿培养(-)。肝肾功能:谷草转氨酶68.1(13~35)U/L,总蛋白89.9(65~85)g/L,球蛋白58.6(20~40)g/L,尿酸 459(155~357)μmol/L,余大致正常。IgG 48.33(6.5~16.0)g/L,IgG1 3650.0(405~1011)mg/dL,IgG2 789.0(169~786)mg/dL,IgG3 335.0(11~85)mg/dL,IgG4 2540.0(3.0~201)mg/dL,总IgE 680(<60)kU/L。血沉 96 mm/h,C反应蛋白 2.71 mg/L。抗核抗体谱:ANA(+),均质型1∶640(<1∶80),胞浆型1∶320(<1∶80),抗ds-DNA(+)1∶40(<1∶10),抗核小体抗体ANuA(+),抗SSA弱阳性,余(-),补体C3 0.366(0.9~1.8)g/L,补体C4 0.187(0.1~0.4)g/L,直接Coombs’试验(2+)。抗髓过氧化物酶抗体(MPO)有反应性(42.8)AU/mL(>24.0),抗中性粒细胞抗体-核周型(p-ANCA)(+)1∶40(<1∶10),抗蛋白酶3(PR3)、抗中性粒细胞抗体-胞浆型(c-ANCA)(-)。狼疮抗凝物、抗心磷脂抗体IgG/IgM、抗β2-糖蛋白抗体(-)。甲状腺功能、肿瘤标志物、T-SPOT.TB、PPD(-),血、尿免疫固定电泳未见单克隆区带。唾液腺超声:左侧颌下腺腺体肿胀,大小约3.9 cm×1.7 cm,回声不均匀减低;右侧颌下腺偏小,2.6 cm×0.8 cm,回声不均匀增强。双侧腮腺腺体肿胀,回声不均匀减低,导管扩张,未见明确占位性病变。腹部超声:肝内低回声结节,胆胰脾肾未见明显异常。肝胆胰脾磁共振成像(MRCP):肝脏S5段病灶,直径约11 mm,肝门区、腹膜后、脾门区多发淋巴结肿大,胰尾部饱满(图2A)。PET/CT示双侧腮腺(SUVmax 4.0)、甲状腺左叶(SUVmax 6.1)、鼻咽顶后壁(SUVmax 3.8)代谢活性增高;肝脏体积增大,脾脏体积增大伴代谢活性弥漫性增高(SUVmax 4.0);胰腺增粗代谢活性增高(SUVmax 2.9);双侧颈部、锁骨上、右肺门、腋窝、腹腔、双侧腹股沟多发大小不等淋巴结、结节显示伴代谢活性增高(SUVmax 7.2);左侧股侧肌内可见低密度影,边界清晰,伴摄取增高(SUVmax 1.8);余未见异常摄取。补充病理免疫组化:左侧下颌肿物:CD38、CD138、IgG(+),IgG4(-);左下颌淋巴结:CD38、CD138、IgG(+),IgG4+/IgG+浆细胞比例约5%;右侧甲状腺肿物:CD38、CD138、IgG(+),IgG4(个别浆细胞+)。
本例患者存在脱发、白细胞减少、ANA(+)、抗ds-DNA(+)、低补体血症、直接Coombs’试验(+),可诊断为SLE,SLE疾病活动指数(SLEDAI)评分10分,为中度活动。另外,患者多个器官存在弥漫性/局限性肿大或团块(甲状腺、腮腺、下颌下腺、胰腺、肝脾、肌肉、淋巴结)伴血清IgG4显著升高(≥135 mg/dL),组织病理学检查示大量淋巴细胞、浆细胞浸润及纤维化改变,但IgG4+/IgG+浆细胞比例<40%且IgG+浆细胞<10个/HPF,可疑诊IgG4-RD。
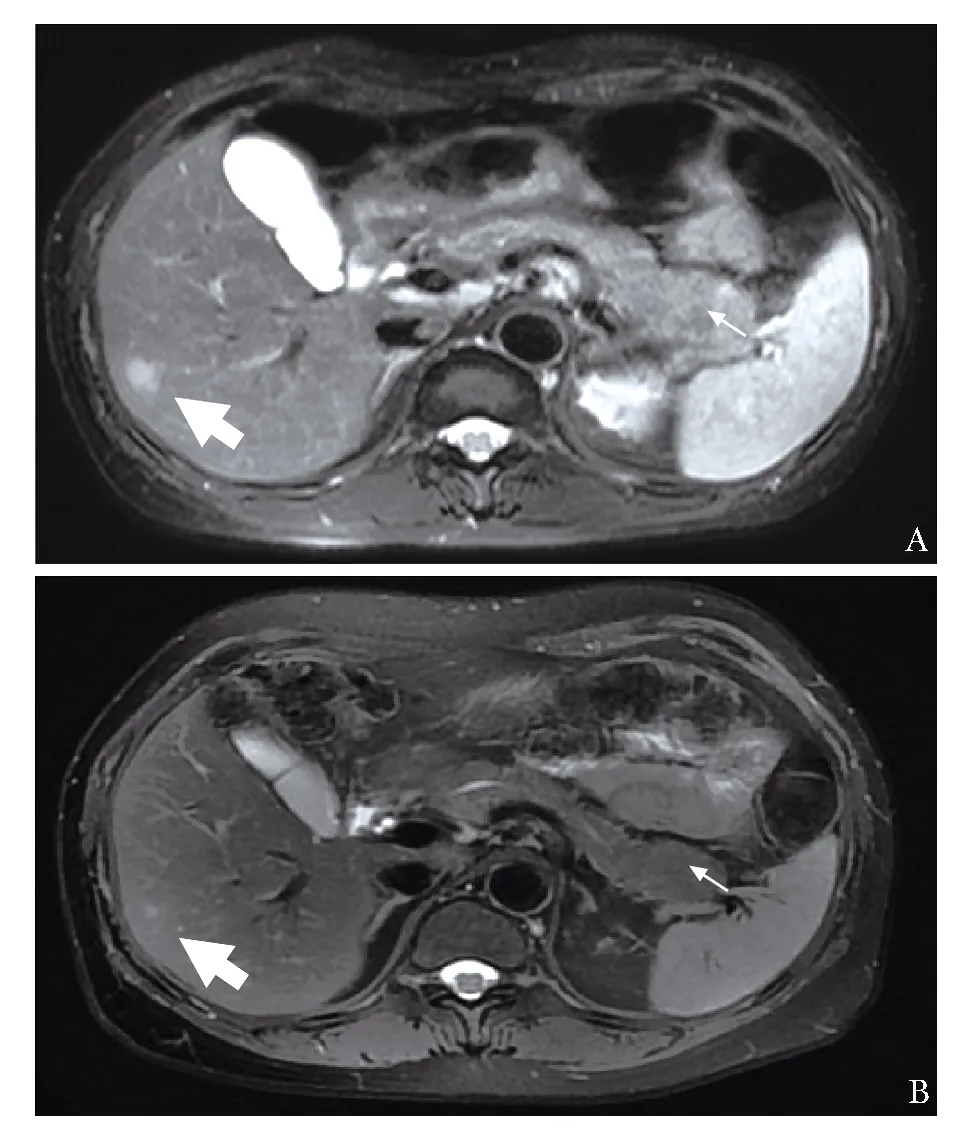
图 2 肝胆胰脾磁共振成像(MRCP) A:肝脏S5段病灶,胰尾部饱满;B:治疗后肝脏S5段病灶缩小,胰尾部饱满减轻
治疗上予口服泼尼松50 mg 每日1次, 3个月后激素规律减量至20 mg每日1次,患者左下颌肿物、左大腿隆起硬结、浅表淋巴结肿大消失,乏力、脱发明显缓解,复查血常规、尿常规、血沉、球蛋白、补体正常,抗dsDNA、MPO、p-ANCA(-),IgG(-),IgG1 1120 mg/dL(405~1011),IgG4 234 mg/dL(5~154),总IgE(-),颈部超声示双侧腮腺基本正常,左侧颌下腺大小正常,右侧颌下腺缩小,双侧颌下腺回声不均,双侧颈部未见淋巴结肿大。MRCP示肝脏S5段病灶、腹腔淋巴结缩小、减少,胰尾部饱满减轻(图2B)。继续规律激素减量,并加用来氟米特(LEF)20 mg 每天1次。9月后激素减量至5 mg每天1次,LEF 20 mg每天1次,复查抗dsDNA抗体 波动于阴性至阳性(1∶10),余血清学指标均无异常。
2 讨论
本例患者兼具SLE和IgG4-RD的临床特征。脱发、白细胞减少、多种自身抗体阳性(ANA、抗ds-DNA、ANuA)、低补体血症、Coombs’试验阳性,符合2012年SLE国际临床协作组(SLICC)关于SLE分类标准[2]。本患者同时有多器官肿大或团块(甲状腺、腮腺、下颌下腺、胰腺、肝脾、肌肉、淋巴结)、血清IgG1、IgG4、IgE升高则为IgG4-RD相关临床表现[3];组织病理学检查示大量淋巴细胞、浆细胞浸润及纤维化改变,但IgG4+/IgG+浆细胞比例<40%,且IgG+浆细胞<10个/HPF,依据2011年提出的IgG4-RD诊断标准,疑诊IgG4-RD[4]。值得注意的是,SLE与IgG4-RD存在许多共同特征,例如SLE可出现IgG4升高、多发淋巴结肿大、脾大,IgG4-RD亦可伴有低补体血症。本例患者甲状腺、下颌下腺、淋巴结组织病理IgG4+浆细胞少见,未见席纹状纤维化、闭塞性静脉炎等IgG4-RD典型病理表现,IgG4-RD诊断存疑,血清IgG4升高、多器官肿大或可为SLE伴随症状。病程较长的IgG4-RD患者组织病理可能以纤维化为主,而缺乏上述典型病理表现[3]。本例患者超声及CT示右侧下颌下腺明显萎缩,左侧下颌下腺病理提示纤维组织增生,可能提示病程较长,或可解释无典型IgG4-RD病理改变。为减少创伤,本次住院期间未进一步获取更多部位组织病理明确诊断;另外,患者否认口眼干表现,未充分排除干燥综合征,为该病例诊断局限之处。
此外,IgG4水平对诊断IgG4-RD特异性、敏感性均欠佳。SLE患者通常伴有IgG4水平下降,特别是在狼疮肾炎的患者中[5]。2015年北京协和医院Zhang等[6]等在分析自身免疫性疾病中IgG亚类水平时发现,100例SLE患者IgG4水平为(57.68±67.98)mg/dL,其中仅有11例IgG4>135 mg/dL,文章未进一步分析IgG4>135 mg/dL患者的临床表现。本例患者IgG4>10 ULN、唾液腺肿大/萎缩、肝脏团块浸润继发于SLE可能性小。2019年ACR/EULAR对IgG4-RD最新的分类标准采用了积分的形式,需首先满足入组标准,不符合排除标准,且积分≥20分可诊断[1]。本例患者有典型的IgG4-RD受累器官,符合入组标准,且血清IgG4≥5 ULN(11分)、≥2副腺体受累(14分)及低补体血症(6分),共计31分>20分。但该患者同时存在多种自身抗体阳性,符合该分类标准的除外标准。由此可见2019年的新标准不适用于考虑IgG4-RD并发其他自身免疫病如SLE的患者。综合考虑,该患者诊断SLE并发IgG4-RD可能,对糖皮质激素治疗反应佳,支持诊断。
SLE并发IgG4-RD的报道并不多见。通过文献复习,检索到了5例SLE并发IgG4-RD可能的病例报道[7-11],特点总结于表1。6例均为亚洲患者,以中老年女性为主,IgG4水平可显著升高或正常;5例有多发淋巴结肿大,2例累及唾液腺;3例胰腺受累,其中2例胰腺肿大,1例自身免疫性胰腺炎;4例有肾脏受累,其中1例为IgG4-RD相关肾脏受累(IgG4-RKD),1例为狼疮肾炎(LN),2例为IgG4-RKD并发LN。本例患者有可疑蛋白尿(160.80 mg/24 h)、脓尿(10~15/HPF),肾功能正常,因患者拒绝未行肾脏穿刺活检。6例患者均对糖皮质激素反应好,其中1例出现病情复发,换用贝利木单抗治疗后好转。
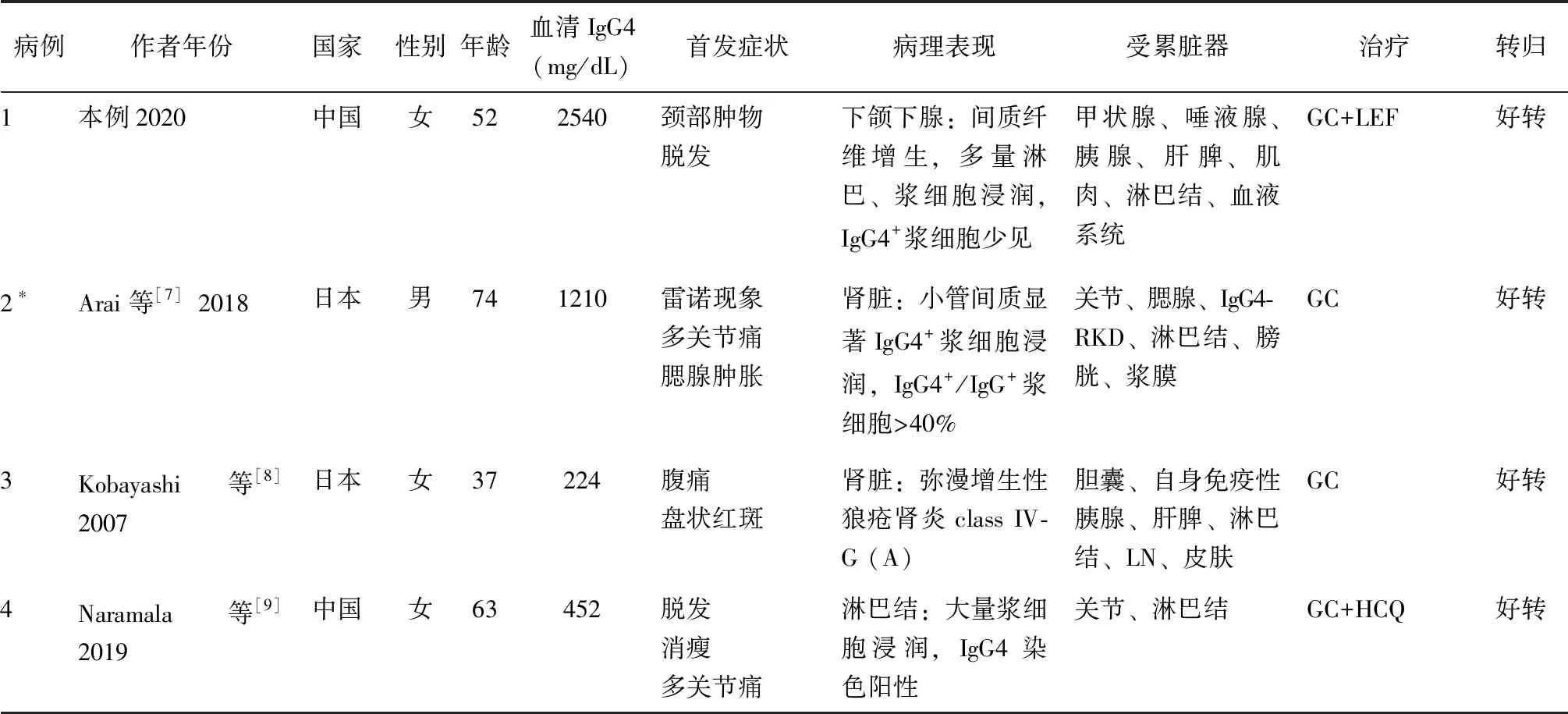
表1 6例SLE并发IgG4-RD患者的临床资料
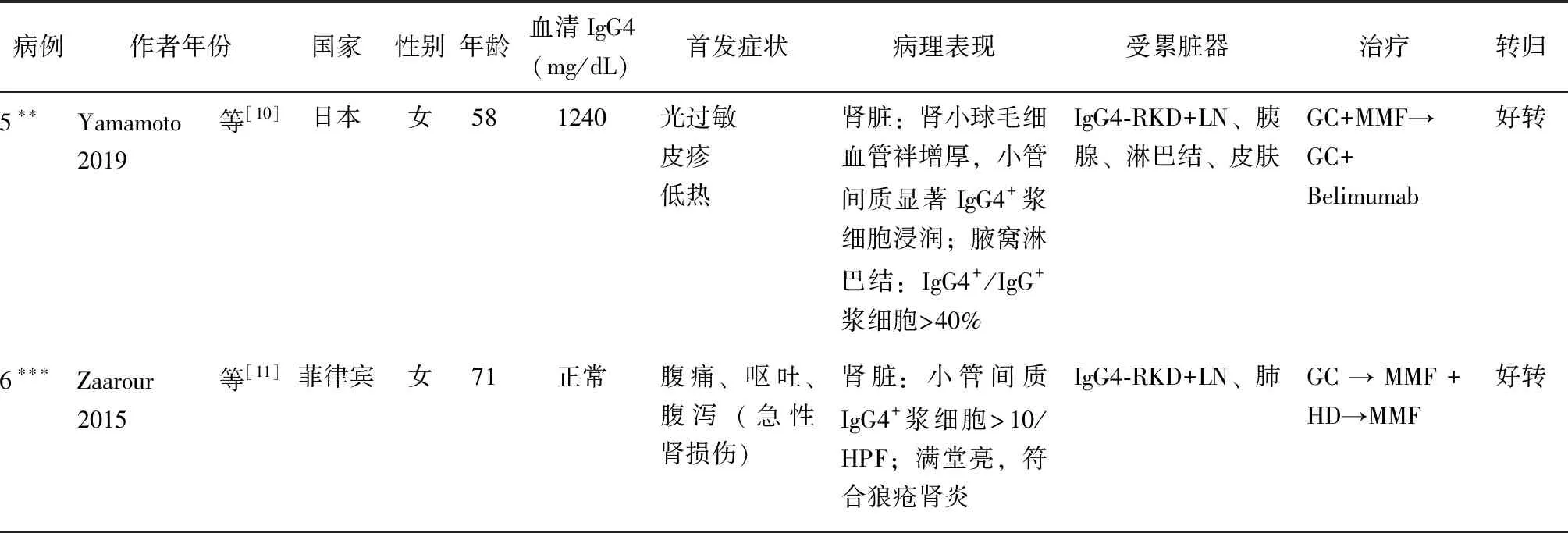
续表1
SLE和IgG4-RD潜在共同的发病机制可能为两者并发出现提供了一定的基础。参与B细胞成熟、自身抗体产生的BAFF和APRIL被认为在IgG4-RD的发病中发挥了关键作用,同样在SLE的发病中也扮演着重要的角色[12-13]。
综上所述,SLE患者可并发IgG4-RD或出现典型的IgG4-RD相关表现,这组患者的发病机制、临床特征、治疗和预后等相关特点需要进一步总结、探讨。
猜你喜欢
当代医药论丛(2022年4期)2022-11-26
检验医学与临床(2022年15期)2022-08-11
中国临床医学(2022年3期)2022-07-08
中国药学药品知识仓库(2022年8期)2022-05-09
临床超声医学杂志(2022年4期)2022-05-07
眼科学报(2022年1期)2022-03-03
中国生物制品学杂志(2022年1期)2022-01-19
临床与实验病理学杂志(2021年7期)2021-09-06
昆明医科大学学报(2021年2期)2021-03-29
医学信息(2021年5期)2021-03-21
