
HPLC法测定枸橼酸莫沙必利分散片的有关物质
2020-11-10 03:50孙亚敏栾会妮徐勤娟李锡勇
山东化工 2020年19期
孙亚敏,栾会妮,徐勤娟,李锡勇
(威海海洋职业学院,山东 威海 264300;)
枸橼酸莫沙必利(Mosapride Citrate)为大日本制药株式会社开发的胃动力药,是一强效选择性5-羟色胺(5-HT4)受体激动剂,通过兴奋肌间神经丛的5-HT4受体,刺激乙酰胆碱释放,从而增强胃肠运动,但不影响胃酸分泌,是一安全有效的胃动力药[1-2]。临床用于慢性胃炎、功能性消化不良、反流性食管炎及手术伴随的一系列胃肠道症状的缓解[3]。本实验参照相关文献[4-5]和JP16版Mosapride Citrate Powder质量标准,进一步优化色谱条件,采用梯度洗脱,建立了检测枸橼酸莫沙必利分散片有关物质的高效液相色谱法,为枸橼酸莫沙必利分散片的质量研究提供了有效的分析手段。
1 实验部分
仪器与试剂
Waters e2695高效液相色谱仪(配有四元泵、在线脱气机、自动进样器、2998 PDA检测器、2489UV检测器、Empower工作站);Mettler Toledo XS205电子天平;Mettler Toledo SevenEasy pH计;SK8200LH数控超声波清洗仪(上海科导超声仪器有限公司)。
枸橼酸莫沙必利对照品(批号:100656-200501,中国药品生物制品检定所),杂质A(批号:7-DPM-9-2,TRC),杂质B(批号:13-SWZ-97-2,TRC),杂质C(批号:4-DPM-73-4,TRC),枸橼酸莫沙必利原料(批号:130301,国内某公司自制),枸橼酸莫沙必利分散片(批号:130801、130802、130803,自制)。所用试剂除乙腈为色谱纯之外,其它均为分析纯。
2 结果与讨论
2.1 色谱条件
色谱柱为Welch SX-C18(4.6×150mm,5μm);以0.05mol/L的枸橼酸溶液(用2mol/L的氢氧化钠溶液调节pH值至4.0)为流动相A,以乙腈为流动相B,按下表梯度程序进行洗脱;检测波长为274nm;柱温为40℃;进样体积为10μl;稀释液为乙腈-水(40∶60)。

表1 色谱条件
2.2 溶液的配制
取枸橼酸莫沙必利分散片细粉适量(约相当于枸橼酸莫沙必利25mg),置25mL量瓶中,加稀释液超声使枸橼酸莫沙必利溶解并稀释至刻度,摇匀,滤过,取续滤液作为供试品溶液;精密量取供试品溶液适量,用稀释液定量稀释200倍,摇匀,作为对照溶液(0.5%)。
2.3 空白辅料
取空白辅料适量(约相当于含枸橼酸莫沙必利20mg),精密称定,置20mL量瓶中,加稀释液超声溶解并稀释至刻度,摇匀,滤过,依法测定,记录色谱图,结果见图1。由结果可知,空白辅料对本品有关物质检查无干扰。
2.4 峰定位
精密称取枸橼酸莫沙必利原料、杂质A、B、C对照品各约5mg,置50mL量瓶中,加稀释液超声溶解并稀释至刻度,摇匀,再精密量取0.5mL,置20mL量瓶中,加稀释液稀释至刻度,摇匀,作为峰定位溶液,依法测定,记录色谱图,结果见图2。结果表明,莫沙必利与相邻杂质及杂质之间的分离度均达到要求。莫沙必利及杂质A、B、C的保留时间(RT)和相对保留时间(RRT)等见表2。
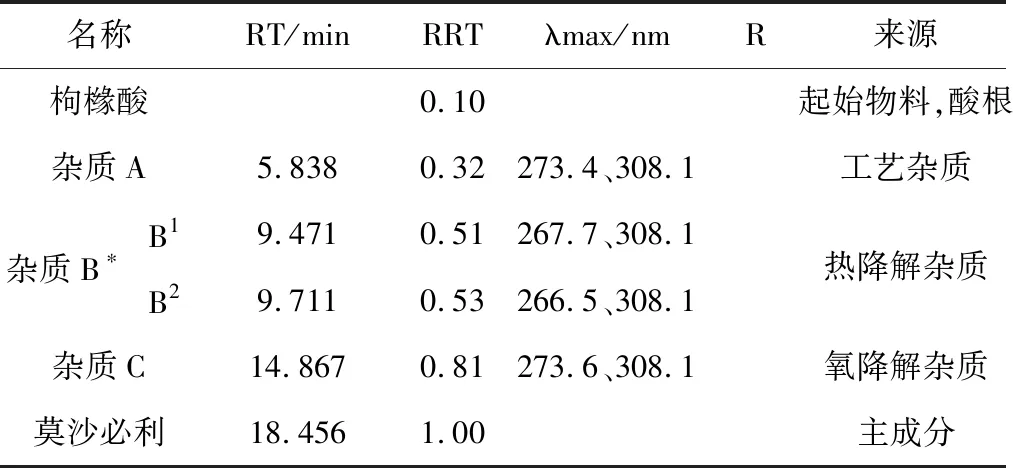
表2 峰定位与系统适用性试验结果Tab.2 Test results of Peak location and system applicabilit
2.5 强制降解试验
按如下方法分别对枸橼酸莫沙必利分散片和空白辅料进行强制降解试验。
2.5.1 酸破坏
取样品细粉适量(约相当于枸橼酸莫沙必利20mg),精密称定,置20mL量瓶中,加稀释液5mL溶解,再加1mol/L-1盐酸溶液5mL,95℃水浴加热30min,冷却,用1mol/L的NaOH溶液5mL中和,用稀释液稀释至刻度,摇匀,滤过。
2.5.2 碱破坏
取样品细粉适量(约相当于枸橼酸莫沙必利20mg),精密称定,置20mL量瓶中,加稀释液5mL溶解,再加1mol/L NaOH溶液5mL,95℃水浴加热30min,冷却,用1mol/L盐酸溶液5mL中和,加稀释液稀释至刻度,摇匀,滤过。
2.5.3 氧化破坏
取样品细粉适量(约相当于枸橼酸莫沙必利20mg),精密称定,置20mL量瓶中,加稀释液5mL溶解,再加3%H2O2溶液5mL,95℃水浴加热10min,冷却,加稀释液稀释至刻度,摇匀,滤过。
2.5.4 高温破坏
取样品细粉适量(约相当于枸橼酸莫沙必利20mg),精密称定,置20mL量瓶中,置105℃烘箱中加热12h,用稀释液超声溶解并稀释至刻度,摇匀,滤过。
2.5.5 光破坏
取在4500Lx光照箱中放置5天样品适量(约相当于枸橼酸莫沙必利20mg),精密称定,置20mL量瓶中,加稀释液超声溶解并稀释至刻度,摇匀,滤过。
精密量取上述破坏溶液,分别依法测定,记录色谱图(图3-图7),考察主成分峰纯度、与降解杂质分离度和降解杂质峰归属(RRT)、最大吸收波长以及物料守恒。结果表明,各强制降解条件下,主峰峰纯度、与相邻杂质的分离度均符合要求;本品对碱、光均不敏感,氧化破坏产生杂质C(莫沙必利N-氧化物);而热破坏主要产生杂质B(莫沙必利柠檬酰胺)、杂质C;主成分及各降解杂质的最大吸收波长均在274nm波长左右;通过计算,各破坏条件下称样量/总峰面积与未破坏时称样量/总峰面积的比值基本一致,且物料守恒。说明本方法能有效的检出各降解杂质,专属性强。

图1 空白辅料Fig. 1 blank excipient
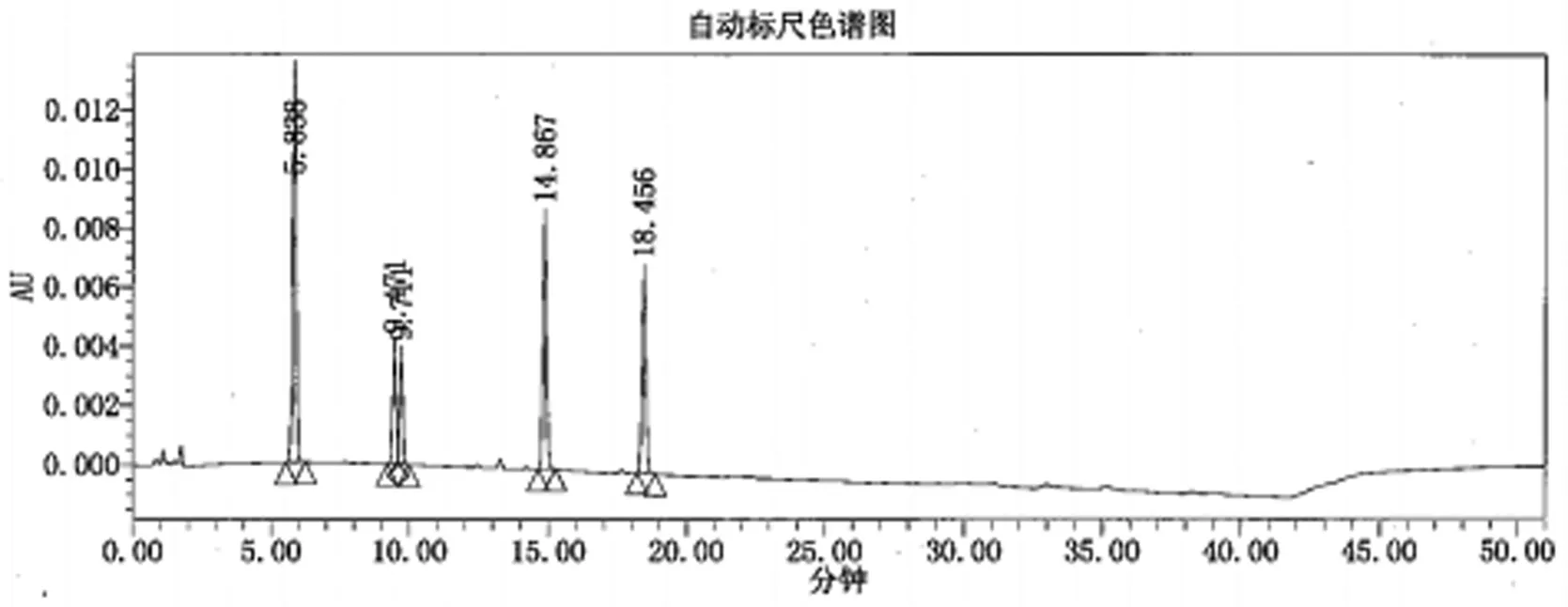
图2 峰定位Fig. 2 Peak location

图3 酸降解Fig. 3 Acid degradation

图4 碱降解Fig. 4 Alkali degradation

图5 氧化降解Fig. 5 Oxidative degradation

图6 高温降解Fig. 6 High temperature degradation
图7 光照降解Fig. 7 Photodegradation
2.6 检测波长的确定
由杂质A、B、C以及强制降解试验中各降解杂质的光谱图可知,在274nm波长处均有较大吸收,而莫沙必利的最大波长为274nm,故选择274nm作为本品有关物质检查的检测波长[6]。
2.7 已知杂质研究
2.7.1 线性关系与范围
取枸橼酸莫沙必利、杂质A、B、C对照品各10.0mg,精密称定,置100mL量瓶中,加稀释液超声溶解并稀释至刻度,摇匀,作为对照品储备液;精密量取0.05mL、0.1mL、0.2mL、0.4mL、0.8mL、1.0mL、1.2mL,分别置20mL量瓶中,加稀释液稀释至刻度,摇匀;精密量取各10μl,分别注入液相色谱仪,记录色谱图。以浓度C(μg/mL-1)为横坐标,峰面积A为纵坐标进行线性回归。结果见表3。

表3 枸橼酸莫沙必利与各杂质的线性关系及检测限与定量限结果Tab.3 Linear relationship , detection limit and quantitative limit results of mosapride citrate and impurities
2.7.2 定量限
以信噪比(S/N)为10∶1的浓度为最低定量限溶液,连续进样6次,记录色谱图。结果表明,莫沙必利和杂质A、B1、B2、C的保留时间的RSD分别为0.07%、0.13%、0.17%、0.20%、0.10%;莫沙必利和杂质A、B、C的峰面积的RSD分别为1.76%、2.09%、2.41%、2.48%。
2.7.3 检测限
以信噪比(S/N)为3∶1的浓度为最低检测限溶液,进样10μl,记录色谱图。结果见表3。
2.7.4 校正因子
采用线性与范围试验项下枸橼酸莫沙必利标准曲线的斜率与各杂质标准曲线的斜率之比计算各杂质相对枸橼酸莫沙必利的校正因子,结果见表4。结果表明,杂质A和杂质B的校正因子均在0.9~1.1范围之外,须用主成分自身对照法计算含量;而杂质C可直接用主成分自身对照法计算含量。

表4 已知杂质校正因子试验结果Tab.4 Test results of known impurity correction factors
2.7.5 精密度
精密量取“2.7.1”项下的对照品储备液1.0mL,置20mL量瓶中,加稀释液稀释至刻度,摇匀,作为100%(5μg/mL-1)浓度溶液。同法配制80%(4μg/mL-1)和120%(6μg/mL-1)浓度溶液,每个浓度配制3份。精密量取上述三种溶液各10μl,,分别注入液相色谱仪,记录色谱图,量取峰面积。结果表明,在80%(4μg/mL-1)、100%(5μg/mL-1)和120%(6μg/mL-1)三个浓度范围内,莫沙必利峰面积的RSD分别为0.89%、0.70%、0.80%,杂质A峰面积的RSD分别为0.77%、0.33%、0.88%,杂质B峰面积的RSD分别为0.84%、0.70%、0.98%,杂质C峰面积的RSD分别为0.72%、0.39%、0.74%。说明本方法重复性较好。
2.7.6 中间精密度
取本品样品(批号:130801)和杂质对照品溶液,由不同试验员在不同的时间用不同仪器,分别依法测定6份,按外标法以峰面积计算已知杂质的含量,计算RSD,考察本方法的重现性。结果表明,杂质A、B、C含量的RSD分别为6.79%、3.15%和5.82%。说明本方法重现性良好。
2.7.7 回收率
取本品适量(约相当于枸橼酸莫沙必利20mg),精密称定,置20mL量瓶中,加入“2.7.1”项下的对照品储备液1.0mL,用稀释液超声溶解并稀释至刻度,摇匀,滤过,作为100%浓度的供试品溶液,同法配制80%浓度的供试品溶液和120%浓度的供试品溶液,每个浓度配制3份;另取“2.7.1”项下的对照品储备液1.0mL,置20mL量瓶中,加溶剂稀释至刻度,摇匀,滤过,作为对照品溶液;另取本品适量(约相当于枸橼酸莫沙必利20mg),精密称定,置20mL量瓶中,用稀释液超声溶解并稀释至刻度,摇匀,滤过,作为空白校正溶液。精密量取供试品溶液、对照品溶液和空白校正溶液各10l,分别注入液相色谱仪,记录色谱图,用空白校正溶液校正后,按外标法以峰面积计算各杂质的含量,计算回收率和相对标准偏差(RSD)。结果表明,杂质A、B、C的平均回收率分别为98.53%、99.01%和101.28%,RSD分别为1.14%、0.98%和1.65%。表明本法的回收率良好。
2.8 溶液稳定性
精密量取对照品溶液和供试品溶液,室温放置,分别于配制后0、2、4、6、8h取样测定,通过对照品峰面积和有关物质(按面积归一化法计算)的变化情况的变化情况,考察溶液的稳定性。结果表明,对照品溶液中莫沙必利和杂质A、B、C峰面积的RSD分别为 0.51%、0.63%、0.60%和0.55%;供试品溶液中杂质A、B、C、其他杂质之和及总杂的含量分别为0.01%、0.07%、0.07%、0.18%和0.33%,基本上没有变化。说明对照品溶液及供试品溶液在8h内稳定。
2.9 耐用性
在本方法色谱条件的基础上,分别改变流速(0.8mL · mi-n和1.2mL · mi-n)、柱温(35℃和45℃)、检测波长(269和279nm)、流动相pH值(3.8和4.2)和色谱柱(Agilent XDB-C18),取供试品溶液和系统适用性溶液,每个条件进样3次,考察本实验的耐用性。结果表明,莫沙必利和杂质A、B、C的保留时间、柱效、对称性和分离度均符合要求,且杂质A、B、C和其他单杂、总杂均无显著性差异(RSD≤10)。
2.10 样品有关物质测定

表5 有关物质检查外标法与自身对照法比较结果Tab.5 Comparison of the results of external standard method and self-control method for related substance examinatio
取本品三批样品(批号:130801、130802、130803),分别采用加校正因子的主成分自身对照法(A法)和外标法(B法)计算样品中已知杂质的含量,结果见表5。结果表明,加校正因子的主成分自身对照法和外标法测定已知杂质结果基本一致,且均符合规定。为方便检测,本实验采用加校正因子的主成分自身对照法检查本品的有关物质。
3 结论
3.1 相对保留时间(RRT)
本方法采用相对保留时间对已知杂质进行定位,通过线性与范围试验、精密度试验和回收率试验中各已知杂质保留时间与莫沙必利保留时间的比值,确定了杂质A、B、C的相对保留时间[7,8];同时,本方法指定了Welch SX-C8色谱柱为本品有关物质检查专用色谱柱。
3.2 校正因子
根据《药品杂质分析指导原则》,杂质的校正因子在“0.9~1.1”之内的,可直接采用主成分的自身对照法对杂质进行定量;超出此范围,而在“0.2~5.0”之内的,可采用加校正因子的主成分的自身对照法对杂质进行定量。故杂质A和B采用加校正因子的自身对照法计算含量,而杂质C可直接采用主成分自身对照法进行定量。
3.3 杂质B
杂质B是一对映异构体(B1和B2),在色谱行为上表现为两个峰。定位时,分别对这这两个峰进行了定位;定量时,则将B1和B2合并为一个峰(杂质B)以峰面积之和进行定量。
3.4 耐用性
本品对波长、柱温、色谱柱的耐用性较好。流速为1.2mL/min时,莫沙必利N-氧化物与相邻的杂质峰合并成一个杂质峰,使杂质总数减少,但总杂没有明显变化,说明增大流速,不利于莫沙必利N-氧化物与相邻杂质峰的分离。流动相pH降低时,主峰保留时间减小,不利于主峰与相邻杂质的分离。
猜你喜欢
基层中医药(2022年7期)2022-11-17
中国兽药杂志(2022年6期)2022-07-04
中国药学药品知识仓库(2022年13期)2022-07-03
中国药学药品知识仓库(2022年7期)2022-05-10
中国典型病例大全(2022年7期)2022-04-22
武警医学(2021年1期)2021-12-05
药品评价(2021年17期)2021-11-06
系统医学(2021年13期)2021-09-28
临床肝胆病杂志(2021年1期)2021-01-26
世界最新医学信息文摘(2020年8期)2020-03-28
