
长链非编码RNA 母系表达基因3调控miR-34a对糖尿病视网膜病变Müller细胞活化及炎症因子分泌的影响△
2021-01-29 03:27王坤朱曼辉陈莉莉涂园园万光明梁申芝
眼科新进展 2021年1期
王坤 朱曼辉 陈莉莉 涂园园 万光明 梁申芝
糖尿病视网膜病变(DR)是糖尿病的常见并发症,也是工作年龄人群致盲的主要原因[1]。虽然DR的确切发病机制尚未完全清楚,但研究表明糖尿病患者视网膜的多种异常均与炎症密切相关,应激性视网膜细胞产生的多种细胞因子失调是导致神经血管损伤的关键因素[2-3],如血管内皮生长因子(VEGF)、白细胞介素-1β(IL-1β)等。其中,Müller细胞源性VEGF是导致DR视网膜炎症、血管渗漏和病理性血管形成的关键因素[4]。尽管抗VEGF治疗具有显著的疗效,但依然存在许多局限性,如需要眼内反复注射,只对疾病晚期有效,更重要的是仅有约一半的患者对治疗有应答[5]。因此,深入探索DR发生发展的机制对于其预防及治疗具有重要的意义。
长链非编码RNA(LncRNA)是一类转录本长度大于200 nt,缺乏蛋白质编码潜能的非编码RNA。随着对LncRNA研究的深入,其在DR中的作用已受到广泛关注[6]。有研究显示,母系表达基因3(maternally expressed gene 3,MEG3)在DR患者血浆及小鼠视网膜中表达降低,沉默MEG3促进视网膜内皮细胞增殖、迁移及管腔形成[7-8],MEG3过表达抑制大鼠视网膜中IL-1β的产生[9]。但是,有关MEG3在DR的Müller细胞中的作用及机制的报道尚少,本研究旨在通过构建的DR小鼠体内模型及高糖刺激视网膜Müller细胞株(MIO-M1)的体外模型,探讨MEG3在DR的Müller细胞活化及炎症因子分泌中的作用及其机制。
1 材料与方法
1.1 材料8周龄健康雄性C57BL/6小鼠30只,体质量20~22 g,购自苏州大学实验动物中心;视网膜Müller干细胞MIO-M1(北纳生物,中国);DMEM培养基、青链霉素、胰蛋白酶、胎牛血清(Gibco,美国);GFAP、VEGF、IL-1β、ELISA试剂盒、辣根过氧化物酶标记的二抗(Abcam,美国);GAPDH单克隆抗体(Proteintech,美国);逆转录试剂盒、实时荧光定量PCR(qRT-PCR)试剂盒、CO2细胞培养箱(Thermo Fisher,美国);MEG3过表达质粒、miR-34a mimic及其阴性对照(RiboBio,广州);Lipofectamine 2000 (Invitrogen,美国);电泳凝胶图像分析系统、聚丙烯酰胺凝胶电泳仪(Bio-Rad,美国);莱卡激光共聚焦显微镜(SP8,德国);冰冻切片机(SLEE,德国);7500实时荧光定量PCR仪(ABI,美国);酶标仪(Gene limited,中国香港)。
1.2 方法
1.2.1 DR小鼠模型制作C57BL/6小鼠共30只饲养于苏州大学SPF级实验动物中心。适应性喂养5 d后,使用随机数字表法将其随机分为两组,分别为正常对照组和DR组,每组各15只。正常对照组小鼠未做处理,DR组小鼠给予高脂饮食喂养8周,再以60 mg·kg-1STZ(美国Sigma公司)连续腹腔注射5 d构建糖尿病模型。当血糖>16.7 mmol·L-1且出现多食、多饮、多尿症状时为造模成功。8周时取各组小鼠双眼眼球及视网膜进行检测。首先通过50 g·L-1戊巴比妥钠(0.01 L·kg-1)腹腔注射麻醉小鼠,其中5只小鼠眼球置于固定液中用于免疫荧光化学检测。剩余10只小鼠眼球置于培养基中,于手术显微镜下沿眼球角巩膜缘剪开,去除角膜、虹膜、晶状体及外层巩膜-脉络膜复合体,取得视网膜,用于后续实验检测。
1.2.2 细胞培养视网膜Müller干细胞MIO-M1培养于含体积分数10%胎牛血清、100 U·mL-1青霉素和100 mg·L-1链霉素的完全培养基中。将细胞置于37 ℃含体积分数5% CO2的培养箱中。2.5 g·L-1胰蛋白酶消化后传代,取对数生长期的细胞用于实验。
1.2.3 细胞转染与处理分别使用5 mmol·L-1(对照组)及30 mmol·L-1(高糖组)葡萄糖刺激MIO-M1细胞24 h用于构建DR体外模型。取对数生长期的MIO-M1细胞接种于6孔板,每孔约2×105个细胞,当细胞融合达60%~80%时,使用Lipofectamine 2000转染MEG3过表达质粒、miR-34a mimic及其阴性对照(NC mimic),继续培养24 h后qRT-PCR检测其转染效率。转染成功后加入含30 mmol·L-1葡萄糖的培养基继续培养24 h,根据转染物的不同将实验分为:pcDNA组、pcDNA-MEG3组、pcDNA+NC mimic组、pcDNA+miR-34a mimic组、pcDNA-MEG3+NC mimic组、pcDNA-MEG3+miR-34a mimic组。取细胞培养上清置于-80 ℃用于酶联免疫吸附实验(enzyme-linked immunosorbent assay,ELISA)检测IL-1β及VEGF蛋白表达。
1.2.4 免疫荧光化学染色快速取出小鼠眼球进行固定,然后分别在100 g·L-1、200 g·L-1和300 g·L-1的蔗糖溶液中脱水过夜。使用冷冻切片机将眼球切成10 μm的薄片,用于免疫荧光化学染色。
对于MIO-M1细胞,将细胞接种在24孔细胞玻片上,并在室温下使用40 g·L-1多聚甲醛固定30 min。随后2 g·L-1Triton X-100室温通透5 min;体积分数5%BSA室温封闭30 min;加GFAP一抗4 ℃孵育过夜,二抗室温避光孵育30 min;DAPI复染后激光共聚焦显微镜观察拍照。
1.2.5 Western blot检测各组小鼠视网膜及细胞中GFAP及VEGF蛋白的表达使用组织及细胞裂解液提取小鼠视网膜组织及MIO-M1细胞总蛋白,BCA法测定蛋白浓度。每组取蛋白质80 μg经过100 g·L-1丙烯酰胺凝胶电泳后使用半干法将蛋白转移至聚偏二氟乙烯膜上,脱脂奶粉室温封闭1 h;加入GFAP及VEGF一抗,4 ℃孵育过夜;加入二抗,37 ℃孵育1 h。最后化学发光法显色,拍照,检测GFAP及VEGF蛋白表达,GAPDH作为内参。
1.2.6 ELISA检测各组小鼠视网膜及细胞上清中VEGF及IL-1β蛋白的表达将小鼠处死后分离视网膜,匀浆、离心吸取的上清和MIO-M1培养基上清各100 μL加入各孔中,按照ELISA 试剂盒说明书进行操作。测量各样本在450 nm波长下的光密度(D)值,获得上清中VEGF及IL-1β蛋白的表达量。
1.2.7 qRT-PCR检测各组小鼠视网膜及细胞中MEG3 mRNA及miR-34a的表达为进一步验证MEG3是否通过下调miR-34a 抑制DR Müller细胞活化及炎症因子的产生,我们将miR-34a mimic及MEG3过表达质粒pcDNA-MEG3分别或同时转染进Müller细胞中,随后高糖刺激24 h。Trizol法提取小鼠视网膜组织及MIO-M1细胞总RNA,逆转录试剂盒将其逆转录为cDNA。加入MEG3及miR-34a特异性引物,PCR反应条件设置为尿嘧啶DNA糖基酶激活50 ℃ 2 min,预变性95 ℃ 2 min,变性95 ℃ 15 s,退火60 ℃ 15 s,延伸72 ℃ 1 min,4 ℃终止反应。每个样本设置三个复孔,使用2-△△Ct法对数据进行相对定量分析。GAPDH作为内参基因。
1.3 统计学分析采用SPSS 22.0对数据进行统计分析,计量资料以均数±标准差表示,各组间比较采用单因素方差分析,两组之间比较采用t检验。检验水准:α=0.05。
2 结果
2.1 DR小鼠视网膜的改变及MEG3 mRNA的表达情况免疫荧光化学染色(图1)及Western blot检

图1 免疫荧光化学染色检测各组小鼠视网膜GFAP表达情况
测(图2A-2B)结果显示,与正常对照组比较,DR组小鼠视网膜中GFAP蛋白表达升高(P<0.01)。ELISA检测(图2C-2D)结果显示,DR组小鼠视网膜VEGF及IL-1β蛋白的表达均增多,差异均有统计学意义(均为P<0.001)。qRT-PCR检测小鼠视网膜中MEG3 mRNA表达(图2E)结果显示,与正常对照组比较,DR组小鼠视网膜中MEG3 mRNA表达减少,差异有统计学意义(P<0.01)。

图2 各组小鼠视网膜GFAP、VEGF、IL-1β蛋白及MEG3 mRNA的表达情况 A、B:Western blot 检测小鼠视网膜中GFAP及VEGF蛋白表达;与正常对照组比较,**P<0.01。C、D:ELISA检测小鼠视网膜VEGF 及IL-1β蛋白表达;与正常对照组比较,***P<0.001。E:qRT-PCR检测小鼠视网膜MEG3 mRNA水平;与正常对照组比较,**P<0.01。
2.2 MEG3对Müller细胞活化及炎症因子产生的影响使用高糖刺激Müller细胞构建DR体外模型检测结果显示,与对照组比较,高糖组Müller细胞中GFAP、VEGF、IL-1β蛋白表达均升高,MEG3 mRNA表达下降,差异均有统计学意义(均为P<0.05),与体内模型检测结果一致。而与高糖组和高糖+pcDNA组比较,高糖+pcDNA-MEG3组可显著增加MEG3 mRNA表达,并抑制高糖诱导的Müller细胞中GFAP、VEGF及IL-1β蛋白表达的升高,差异均有统计学意义(均为P<0.05)(图3-图4)。
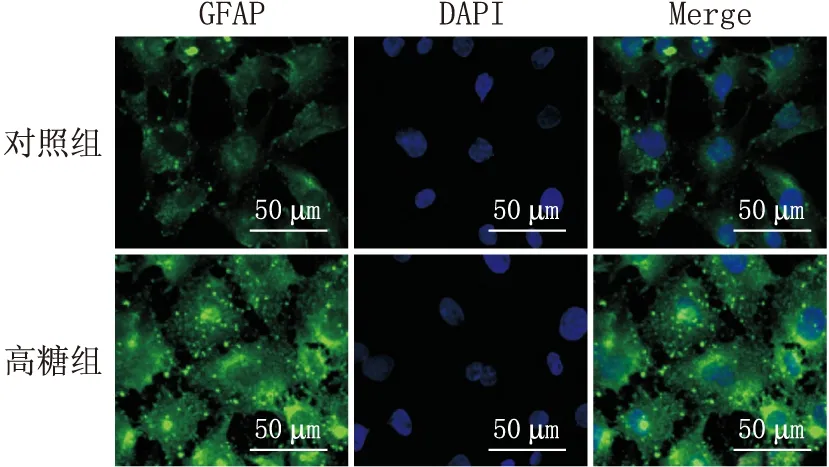
图3 免疫荧光化学染色检测Müller细胞中GFAP表达

图4 MEG3对Müller细胞活化及炎症因子产生的影响 A:qRT-PCR检测MEG3 mRNA水平,与对照组比较,**P<0.01;与高糖组比较,#P<0.05。B、C:Western blot检测Müller细胞中GFAP及VEGF蛋白表达;与对照组比较,**P<0.01;与高糖组比较,#P<0.05。D、E:ELISA检测VEGF蛋白及IL-1β蛋白表达;与对照组比较,***P<0.001;与高糖组比较,##P<0.01。
2.3 MEG3调控miR-34a对Müller细胞活化及炎症因子产生的影响与pcDNA+NC mimic组比较,pcDNA+miR-34a mimic组GFAP、VEGF及IL-1β蛋白表达均增加,而其在pcDNA-MEG3+NC mimic组中表达均降低,差异均有统计学意义(均为P<0.05)。与pcDNA-MEG3+NC mimic组比较,pcDNA-MEG3+miR-34a mimic组中GFAP、VEGF及IL-1β蛋白表达均升高,差异均有统计学意义(均为P<0.05)(见图5)。
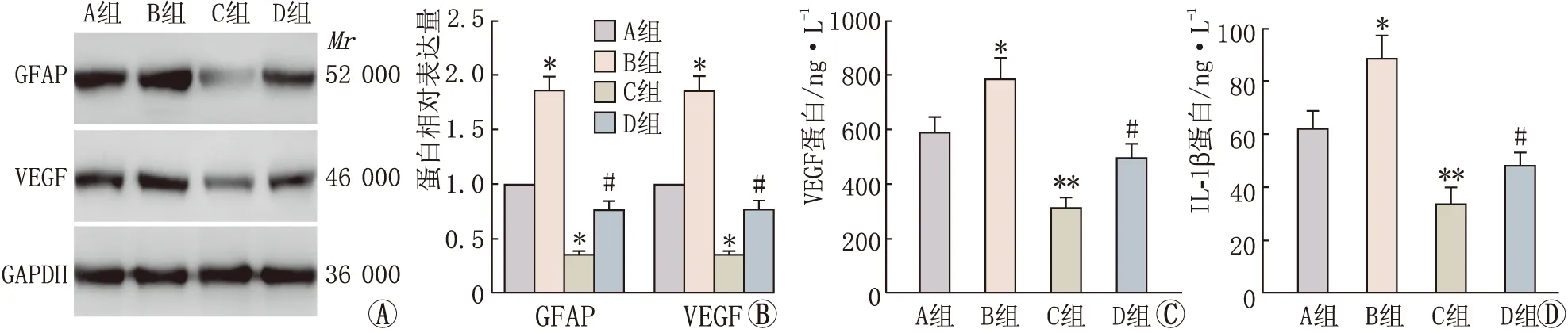
图5 MEG3调控miR-34a对Müller细胞活化及炎症因子产生的影响 A、B:Western blot 检测Müller细胞中GFAP及VEGF蛋白表达;与pcDNA + NC mimic组比较,*P<0.05;与pcDNA-MEG3+NC mimic组比较,#P<0.05。C、D:ELISA检测VEGF蛋白及IL-1β蛋白表达;与pcDNA + NC mimic组比较,*P<0.05,**P<0.01;与pcDNA-MEG3+NC mimic组比较,#P<0.05。A组:pcDNA+NC mimic组;B组:pcDNA+miR-34a mimic组;C组:pcDNA-MEG3+NC mimic组;D组:pcDNA-MEG3+miR-34a mimic组。
2.4 MEG3对miR-34a的调控作用qRT-PCR检测小鼠视网膜及Müller细胞中miR-34a的表达,结果显示,与正常对照组比较,DR组小鼠视网膜中miR-34a mRNA表达升高(图6A),差异有统计学意义(P<0.05)。与体内实验结果一致,体外实验结果显示,与对照组Müller细胞比较,高糖组Müller细胞中miR-34a mRNA表达亦升高(图6B),差异有统计学意义(P<0.05);转染MEG3过表达质粒后,与pcDNA组比较,pcDNA-MEG3组中miR-34a mRNA表达降低(图6C),差异有统计学意义(P<0.05)。

图6 MEG3对miR-34a的调控作用 A:qRT-PCR检测小鼠视网膜中miR-34a水平;与正常对照组比较,**P<0.01。B:qRT-PCR检测Müller细胞中miR-34a水平;与对照组比较,**P<0.01。C:正常条件下转染MEG3过表达质粒后24 h,qRT-PCR检测Müller细胞中miR-34a水平;与pcDNA组比较,**P<0.01。
3 讨论
DR是糖尿病引起的最严重的眼部并发症之一。DR早期一般无症状,但当患者开始出现视力障碍时,视网膜病变已发展到几乎不可逆转的程度[10]。DR主要的病理特点是微血管病变,但在微血管发生改变之前,即出现视网膜神经元及胶质细胞病变[11-12]。Müller细胞为神经视网膜中主要的胶质细胞,其功能障碍被认为是DR发生发展的关键因素[13]。
Müller细胞是视网膜中主要的胶质细胞。它是跨越整个视网膜的唯一细胞,并且与视网膜血管和神经元都紧密接触[14]。在DR早期,Müller细胞即被激活并分泌多种细胞因子,参与视网膜细胞的免疫和炎症反应[13,15]。Müller细胞被激活最明显的标志之一是GFAP的表达增加[16]。GFAP是反应性胶质增生的常见标志物,正常条件下Müller细胞几乎不表达GFAP[17]。本研究中,DR小鼠视网膜中GFAP表达升高,表明Müller细胞被激活。Bai等[4]通过条件性敲除小鼠Müller细胞中VEGF,发现阻断Müller细胞源性VEGF可以显著抑制缺血诱导的视网膜新生血管和血管渗漏,并减少缺血诱导的血-视网膜屏障破坏。提示Müller细胞源性的VEGF是导致DR视网膜炎症、血管渗漏和病理性血管形成的关键因素。本研究中,通过免疫荧光化学染色及Western blot检测,我们发现DR小鼠视网膜及高糖刺激的Müller细胞中,VEGF蛋白表达均升高,与以往研究结果一致。此外,炎症因子IL-1β的分泌亦增多。通过过表达MEG3抑制了Müller细胞的激活及VEGF和IL-1β的产生。提示DR早期Müller细胞处于激活状态,抑制Müller细胞的活化可能成为DR的重要治疗策略。
近年来,大量证据表明MEG3在DR中发挥重要作用[7-9]。Zhang等[7]研究发现,DR患者及糖尿病非DR患者血清中MEG3较正常对照组表达下降,IL-1β及转化生长因子-β1表达升高。提高视网膜色素上皮(RPE)细胞中MEG3的表达水平,减少了高糖刺激引起的IL-1β和转化生长因子-β1的升高。DR大鼠中,MEG3通过抑制插头样转录因子O1及IL-1β改善糖尿病大鼠视网膜病变[9]。沉默MEG3加重了糖尿病引起的体内微血管功能障碍,并在体外促进了内皮细胞的增殖、迁移和管腔形成功能[8]。但MEG3在视网膜Müller细胞中的作用报道尚少。本研究发现,高糖处理的Müller细胞中MEG3 mRNA表达下降,过表达MEG3 mRNA减少Müller细胞的活化及VEGF、IL-1β的释放,过表达MEG3 mRNA或许可以作为DR的潜在治疗靶点,抑制DR的发生发展。
LncRNA能够识别microRNA (miRNA)的互补序列,并与其靶向结合。miRNA是一类普遍存在于生物体基因组中的由内源基因编码的单链非编码RNA分子,长度约为22个核苷酸。随着对miRNA研究的深入,有证据表明miRNA在DR发生发展中发挥重要作用[18]。miR-34a在高糖刺激的RPE细胞中表达升高,沉默miR-34a可抑制高糖导致的RPE细胞活力降低及凋亡增加[19]。本研究中,DR小鼠视网膜及高糖处理的Müller细胞中miR-34a表达均升高,过表达miR-34a促进了高糖诱导的Müller细胞中GFAP及VEGF、IL-1β蛋白的表达。Huang等[20]证实,MEG3充当miR-34a的竞争内源性RNA,并以Argonaute 2依赖性方式相互抑制。高糖处理的RPE细胞中,MEG3通过抑制miR-34a,抑制RPE细胞的炎症与凋亡[19]。但有关Müller细胞中两者的靶向关系报道尚少。本研究结果发现,MEG3 mRNA在DR小鼠视网膜及高糖刺激的Müller细胞中表达均降低,且过表达MEG3 mRNA后miR-34a表达降低,提示MEG3对miR-34a具有调节作用。
为进一步验证MEG3是否通过下调miR-34a抑制DR中Müller细胞活化及炎症因子的产生,我们将miR-34a mimic及MEG3过表达质粒pcDNA-MEG3分别或同时转染进Müller细胞中。结果显示,pcDNA-MEG3组GFAP、VEGF及IL-1β蛋白表达均减少。相反,miR-34a mimic组转染后细胞内GFAP、VEGF及IL-1β蛋白表达均增多。共转染pcDNA-MEG3及miR-34a mimic 后,GFAP的表达及炎症因子的分泌较单独过表达MEG3组增多。提示MEG3 通过下调miR-34a 抑制DR中Müller细胞活化及炎症因子的产生。
综上所述,本研究结果表明,MEG3 mRNA在DR小鼠视网膜及高糖刺激的Müller细胞中表达均下降,MEG3过表达可抑制Müller细胞的活化及VEGF、IL-1β的释放,其机制与调控miR-34a有关。本研究对MEG3在视网膜Müller细胞中的生物学功能所做的基础性研究,将为DR发病机制的阐明及新治疗手段的开发提供一定的理论基础。
猜你喜欢
现代仪器与医疗(2022年2期)2022-08-11
材料与冶金学报(2022年2期)2022-08-10
中医眼耳鼻喉杂志(2021年1期)2021-07-22
中医眼耳鼻喉杂志(2021年2期)2021-07-21
云南化工(2020年11期)2021-01-14
作文成功之路·小学版(2020年6期)2020-07-27
中成药(2018年6期)2018-07-11
中成药(2017年8期)2017-11-22
湖南中医药大学学报(2016年1期)2016-12-01
长江蔬菜(2015年3期)2015-03-11
