
LC-MS/MS测定风味发酵乳中的矢车菊素-3-葡萄糖苷和矢车菊素-3-芸香糖苷
2021-02-03 11:47*李丽
当代化工研究 2021年24期
*李 丽
(江苏联合职业技术学院镇江分院 镇江高等职业技术学校 江苏 212000)
引言
矢车菊素-3-葡萄糖苷(盐酸盐分子式C21H21O11Cl,CAS:7084-24-4,图1)和矢车菊素-3-芸香糖苷(盐酸盐分子式C27H31O15Cl,CAS:18719-76-1,图2)属于黄酮类物质,在黑加仑、桑椹等植物产品中广泛存在[1]。
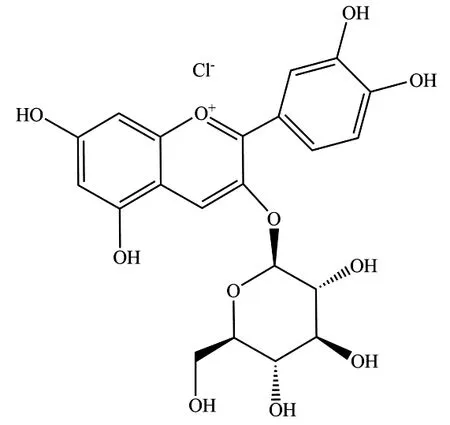
图1 矢车菊素-3-葡萄糖苷盐酸盐
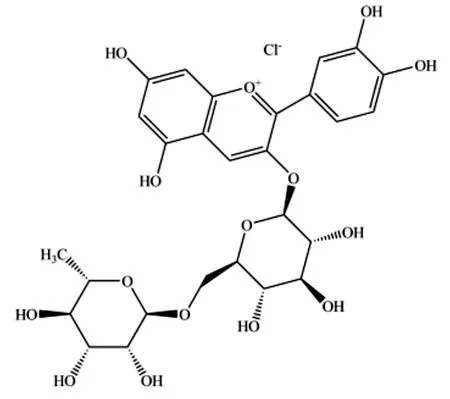
图2 矢车菊素-3-芸香糖苷盐酸盐
对于桑椹红作为食品着色剂的使用,GB1886.345-2021《食品安全国家标准 食品添加剂 桑椹红》[2]给出了两种分子结构式:矢车菊素-3-葡萄糖苷,[C21H21O11]+X-;矢车菊素-3-芸香糖苷,[C27H31O15]+X-,其中X-为平衡离子。虽然C3G、C3R具有良好的健康功效[3],但是在GB2760-2014《食品安全国家标准食品添加剂使用标准》[4]的A.2表中,未将其列入可在各类食品中按生产需要适量使用的食品添加剂范畴。A.1表只规定了果糕类、糖果等六类食品中允许添加桑椹红色素。日常食用的风味发酵乳并没有在允许使用的品种之列。
目前,C3G和C3R的检测有高效液相色谱法(HPLC)[5-7]和LC-MS/MS法。HPLC法属于常规的定量方法,关注的是草莓、黑加仑等产品中这两种化合物的含量,C3G和C3R的浓度相对较高易于测定。但HPLC测定C3G和C3R存在以下的缺陷:(1)参考国际通行的做法,对于禁用物质的检测,液相色谱的测定结果不易作为最终的判定依据,需要配合质谱、红外、拉曼、核磁等技术手段对分子结构进行定性或者更换不同的色谱柱予以确认。(2)样品前处理的方法或者色谱分离条件不适用于质谱的分析。在上述HPLC方法中,样品待测液中含有高浓度的无机酸(如磷酸)或者流动相中使用了盐酸,无机酸的存在会严重污染或者损害质谱检测器。Janine M Cooney[8]等采用直接过SPE柱的方式,用LCMS/MS测定了尿液中C3G和C3R的含量,样品基质相对简单,易于处理;Chunxia Yang等用LC-MS/MS测定了血清中C3G的含量,在样品前处理过程中待测组分的损失较大,需要用槲皮素-3-葡萄糖苷作为内标进行校正,该方法能否适用风味发酵乳的测定尚待评估。
针对风味发酵乳中含有蛋白、糖、甜味剂、增稠剂、乳化剂等物质的特点,本研究拟采用乙腈、甲醇、乙酸铅、三氯乙酸等不同的溶液提取待测组分,并对其提取效率和净化方法进行评估和确认,为风味发酵乳中C3G和C3R的定性定量测定提供了技术方面的参考。
1.材料与方法
(1)仪器及试剂
Thermo TSQ Quantan Access液相色谱-串接质谱仪(配电喷雾离子源(ESI)和自动进样器,美国Finnigen公司);XPE205电子天平(瑞士梅特勒公司);3K15高速离心机(德国Sigma公司)。
甲醇、乙醇、正己烷(色谱纯,德国Merck公司);甲酸、氨水、乙酸铅、三氯乙酸(分析纯,上海国药集团);实验用水采用高纯水(美国Millipore公司);水相膜(0.22μm,上海安谱公司);Poly-Sery MCX固相萃取柱(500mg 6mL,上海安谱公司);C3G标准品(CAS 7084-24-4)、C3R标准品(CAS 18719-76-1)(纯度均大于≥98.0%,成都草源康生物科技有限公司)。
(2)实验方法
①标准溶液的配置
称取C3G和C3R标准品各5.0mg(±0.1mg)于15mL棕色玻璃瓶中,加入10.0mL水(含0.4%甲酸)溶解振荡,配制成浓度为500mg/L的标储。冷冻(-18℃)密闭暗室保存。根据后续需要加0.2%甲酸水稀释。
②制样
取同一批次的风味发酵乳4盒,用组织捣碎机混匀,待用。
③样品前处理过程
A.提取待测组分。称样2.00g(±1.0mg)于15mL具塞离心管中,加10mL 10%三氯乙酸溶液涡旋10s,超声处理8min。冷冻离心25min(8000r/min,R=8cm,-4℃),取上清液加3ml正己烷涡旋1min,离心3min,弃去正己烷层,水相待净化。
B.净化。Poly-Sery MCX柱先后用5mL甲醇、5mL 0.2%甲酸水清洗,保持柱床的湿润。加入5mL提取液过柱。用5mL甲醇和5mL 0.2%甲酸水溶液清洗柱床。用洗耳球将柱床吹干,加入8mL 15%氨水-甲醇洗脱,收集全部滤液氮吹至200μL左右,补加0.4%甲酸水至2.0mL,采用涡旋的方法再次溶解,过水相膜待测定。
(3)液相色谱-串接质谱检测条件
①液相色谱条件
Agilent Eclipse Plus C18柱(150mm×2.1mm,3.5μm);流动相C:乙醇(含0.05%甲酸),流动相D:水(含0.2%甲酸),流动相A:甲醇,梯度洗脱见表1。流速0.25mL/min;进样量20μL;室温。
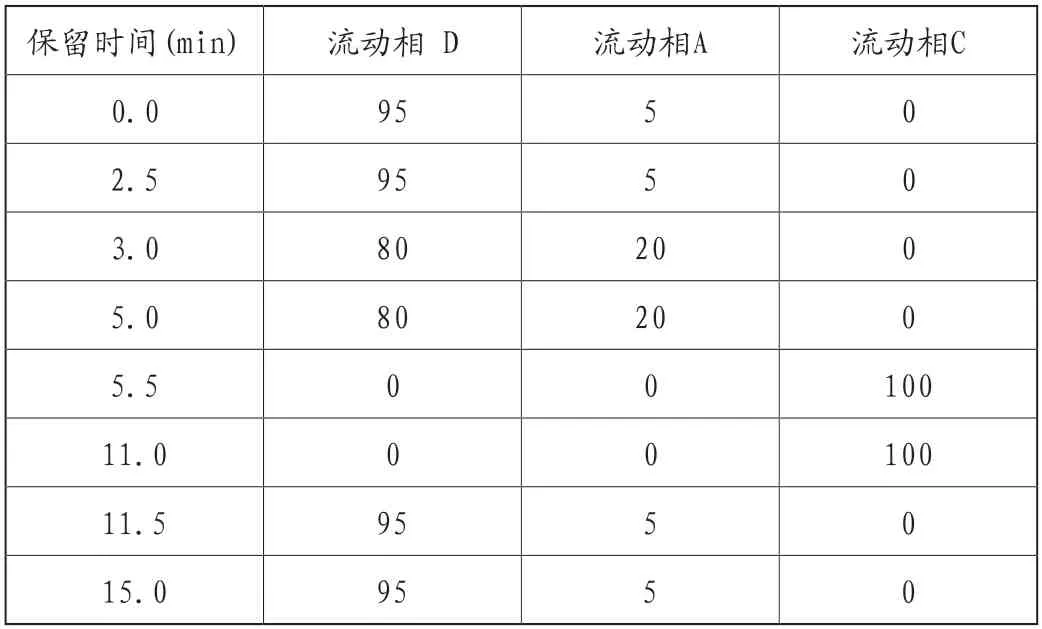
表1 色谱梯度洗脱条件
②质谱条件
电喷雾离子源(ESI),正离子模式扫描;质谱扫描方式:多反应监控(MRM);喷雾电压3800V;传输毛细管温度400℃;辅助气流量2.5mL/min;壳气流量10mL/min。
2.结果与分析
(1)质谱条件的优化

续表
将标准溶液配成10.0mg/L的浓度,在正离子模式下进行母离子、子离子的优化。C3G、C3R的监测量离子对、能量、保留时间见表2。标液的总离子流图和质谱见(图3、图4)。
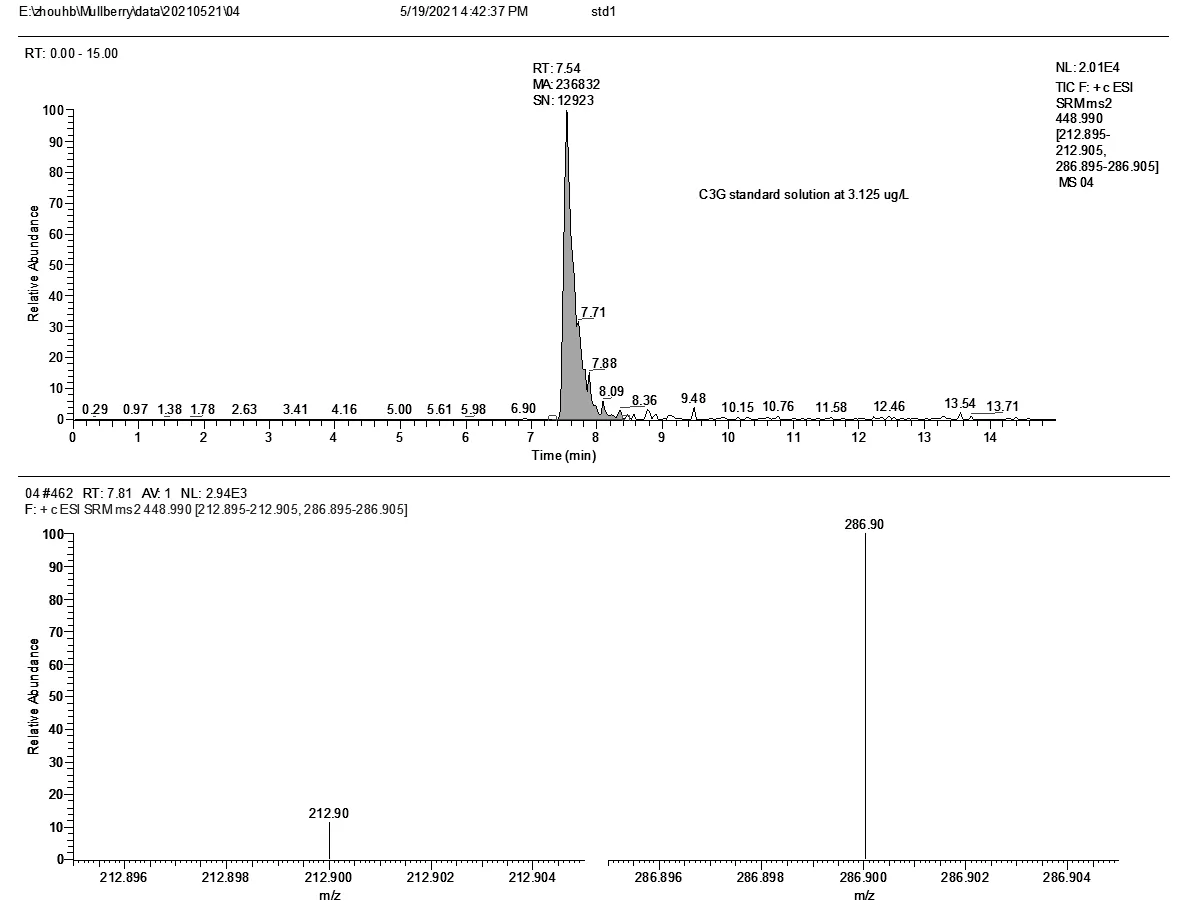
图3 C3G的总离子流图和质谱图(浓度3.125μg/L)
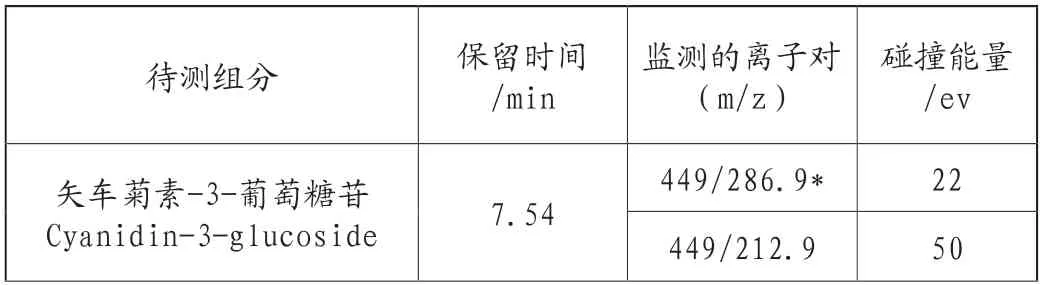
表2 待测组分的质谱参数、保留时间
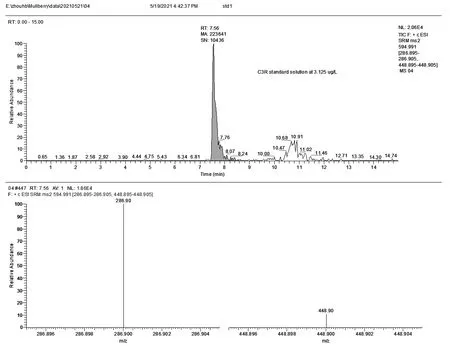
图4 C3R的总离子流图和质谱图(浓度3.125μg/L)
(2)样品提取和净化条件的选择
①在空白样品中加入相同浓度的混合标准溶液情况下(50μg/kg),考察了0.1%甲酸乙腈、0.1%甲酸乙腈+甲醇(乙腈:甲醇=2:1,v/v)、20%乙酸铅、三氯乙酸作为常用的蛋白沉淀剂的沉淀效果以及提取效率。用0.1%乙腈提取待测组分,沉淀效果好、上清液澄清,但是C3G、C3R的回收率分别只有63.1%和60.2%;用0.1%乙腈+甲醇提取待测组分,C3G、C3R的回收率分别为44.1%和25.2%;20%乙酸铅的提取效率相对较差,C3G、C3R的回收率分别只有3.58%和4.52%,可能跟高浓度的乙酸铅中的离子占据了MCX的吸附点位有关。低浓度的三氯乙酸沉淀效果不佳,会产生比较大基质干扰,回收率偏高。经综合测试,采用10%的三氯乙酸处理样品可以取得良好的效果。同等浓度的C3G加标测定值的峰面积大小顺序依次为:10%的三氯乙酸>乙腈>0.1%甲酸乙腈+甲醇>20%乙酸铅(图5);C3R的测定结果与C3G一致(图6)。
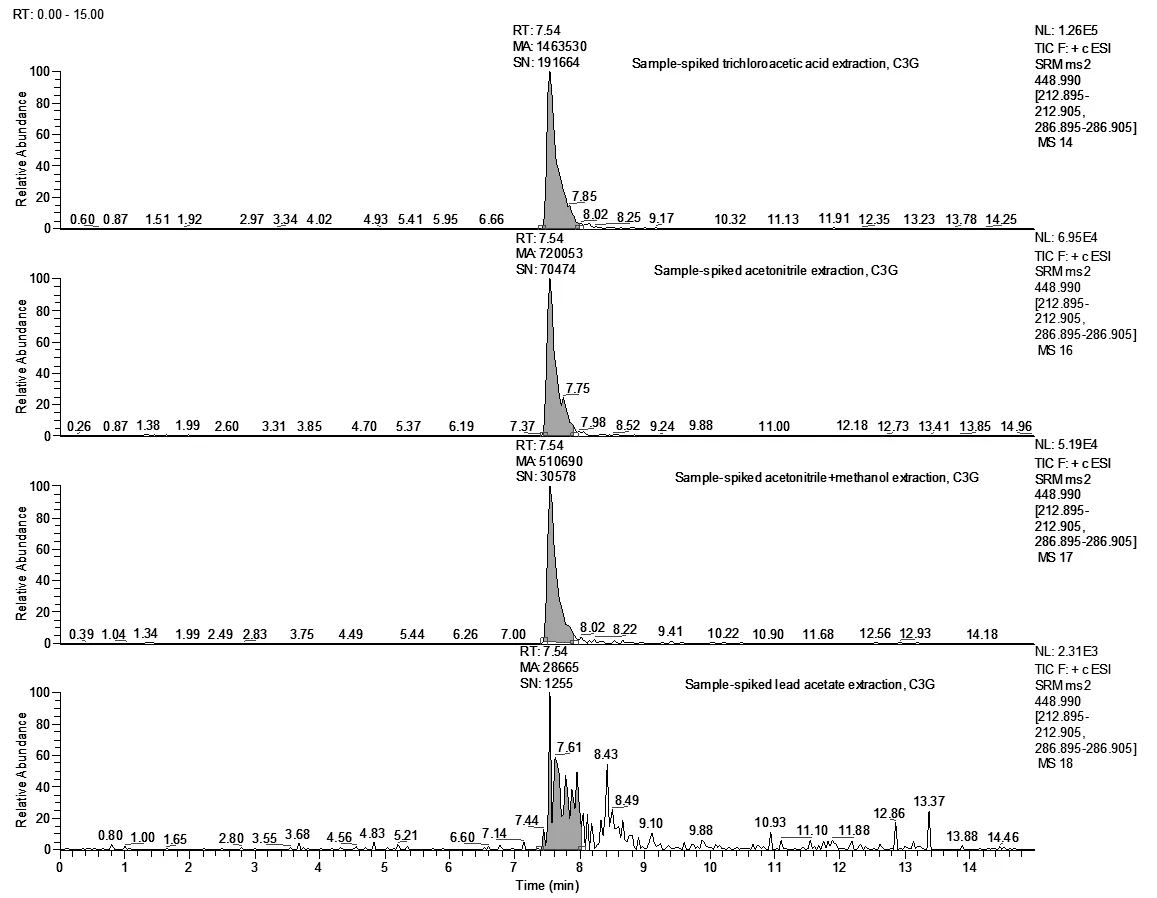
图5 四种溶剂处理加标样品后,C3G的总离子流图
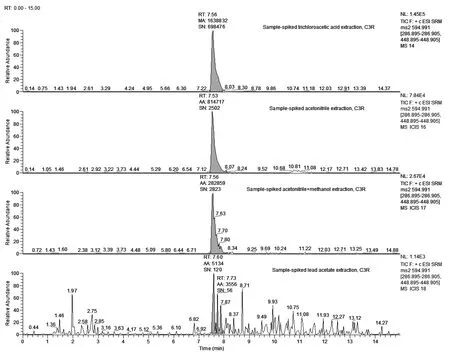
图6 四种溶剂处理加标样品后,C3R的总离子流图
②在色谱的分离阶段,选择了甲醇、0.05%甲酸甲醇、乙醇、0.05%甲酸乙醇作为洗脱溶液。试验中发现用甲醇洗脱C3G和C3R,会产生少量的柱残留。采用0.05%甲酸乙醇洗脱待测组分,不仅可以消除柱残留现象,也有利于待测组分响应值的提高。
(3)方法的标准曲线、线性范围、检出限和定量限
吸取100μL标准混合溶液(1000μg/L)于900μL 0.2%甲酸水溶液中,涡旋混匀。再将混合溶液用0.2%甲酸水倍比稀释至6个浓度:3.12μg/L、6.25μg/L、12.5μg/L、25.0μg/L、50.0μg/L、100μg/L。以定量离子的峰面积为横坐标,质量浓度为纵坐标,绘制工作曲线。所得工作曲线线性良好,相关系数R2大于0.995(图7、图8)。
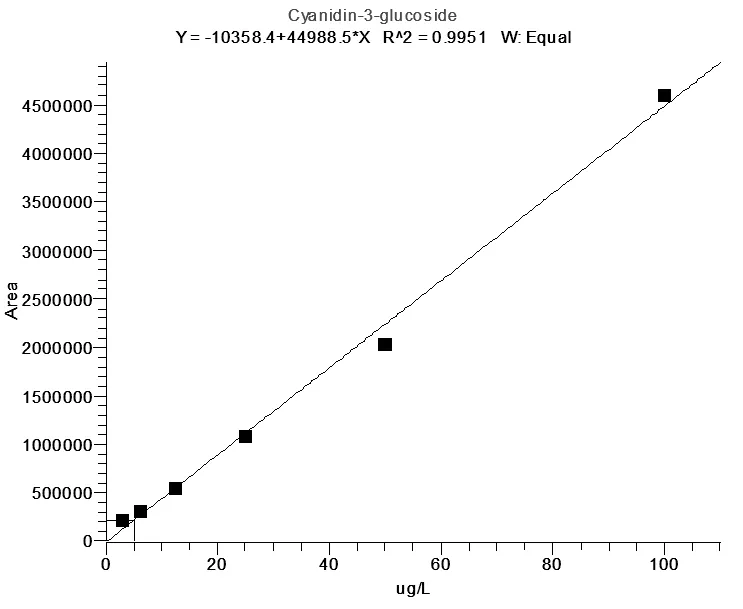
图7 C3G标准曲线图(浓度3.12μg/L-100μg/L)
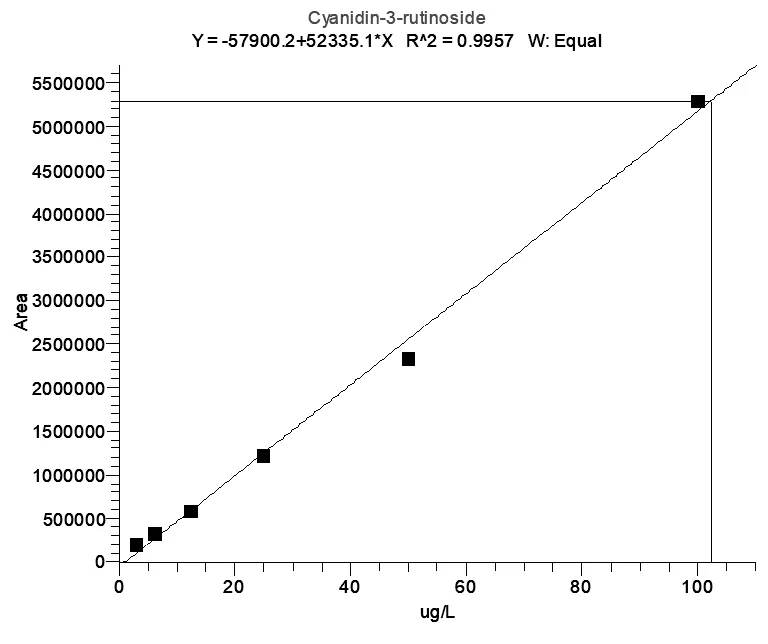
图8 C3R标准曲线图(浓度3.12μg/L-100μg/L)
参照Eurachem的方法[8],计算出这两种化合物检测方法的检出限和定量限均为2.50μg/kg、5.00μg/kg。
(4)回收率及精密度
空白样品进行加标回收检测,添加水平:5.00μg/kg(LOQ)、10.0μg/kg(2×LOQ)、50.0μg/kg(10×LOQ),每个平行测定6次。C3G的加标回收率为103%~115%,相对标准偏差(RSDs)为4.17%~5.88%;C3R的加标回收率为105%~119%,相对标准偏差5.32%~6.07%。空白样品、加标样品总离子流见(图9、图10)。
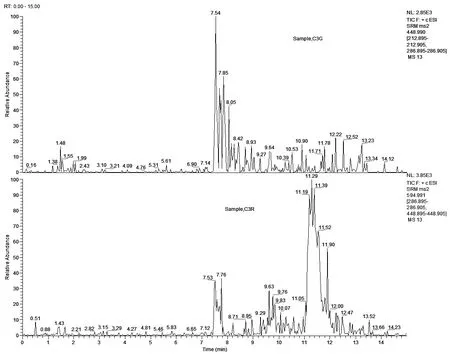
图9 空白样品的总离子流图
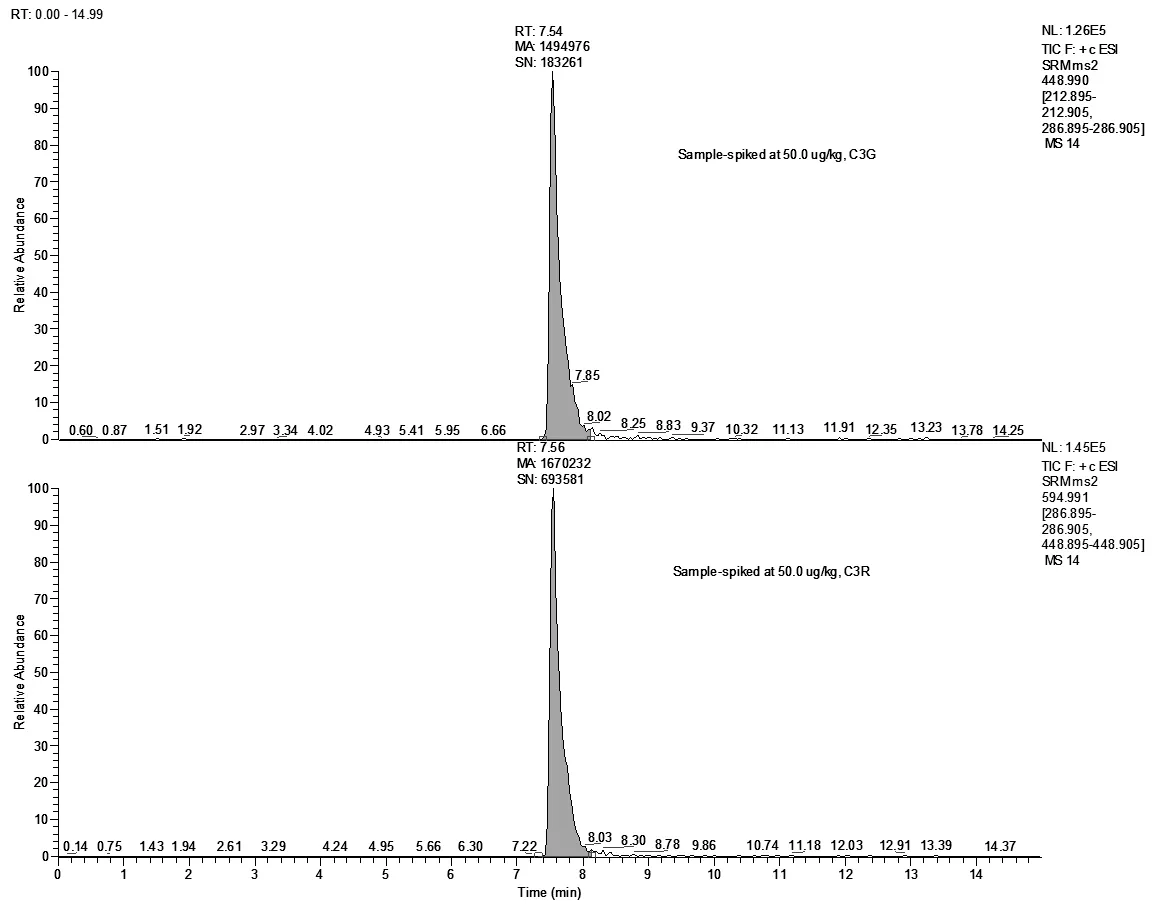
图10 发酵乳加标样品的总离子流图(加标浓度50.0μg/kg)
(5)样品的筛查
原味、红枣味等8份从随机采购的发酵中都没有检出。
3.结论
风味发酵乳经三氯乙酸沉淀蛋白并提取纯化,把色谱洗脱条件及仪器参数进行调整优化后,建立了液-质连用检测C3G、C3R的检测方案,应用于市面上常见的发酵乳的检测。所测的8份随机购买的发酵乳,都没有检测出上述两种物质。该方法的建立为将来更加合理、科学地制定C3G、C3R的使用标准,防止该类色素的不规范使用,提供了技术支撑。
猜你喜欢
学苑创造·A版(2021年4期)2021-04-18
小学阅读指南·低年级版(2021年3期)2021-03-19
江苏农业科学(2021年1期)2021-03-15
理化检验-化学分册(2021年1期)2021-03-06
井冈山大学学报(自然科学版)(2021年1期)2021-03-05
电子科技大学学报(2017年6期)2017-12-22
河北电力技术(2017年4期)2017-09-25
科技传播(2016年15期)2016-11-30
中西医结合心脑血管病杂志(2016年1期)2016-01-25
中国氯碱(2014年11期)2014-02-28
