
利用二维流体分子热力学模型计算气体混合物在固体界面的吸附等温线
2021-03-06 02:58陈博亚李明宴朱雨航彭昌军刘洪来
化工学报 2021年2期
陈博亚,李明宴,朱雨航,彭昌军,刘洪来
(华东理工大学化学与分子工程学院,上海200237)
引 言
吸附分离被认为是一种更具经济效益的气体分离与净化手段。页岩气中甲烷/乙烷和甲烷/二氧化碳的分离[1−4]、天然气和沼气中降低二氧化碳和氮气的占比等[5−6]都可采用气固吸附来实现。近些年来,一些吸附性强、选择性高的多孔固体材料已在气体捕集与分离中受到广泛关注[7−10]。在吸附分离中,吸附量的测定除采用实验外,也可采用纯经验模型和具有一定理论基础的热力学模型进行关联和预测,如扩展的朗缪尔模型(EL−A)[11]、理想吸附溶液模型(IAST)[12]、Wilson 空穴溶液模型(W−VST)[13−15]、二维流体状态方程(2D−EOS)[16]和二维变阱宽微扰链统计缔合流体理论(SAFT−VR−2D)[17]等。但目前的模型或是有特定的应用范围[18],或是含有较多经验参数[19],或是不能较好应用于气体混合物[20]。
实际上,吸附于固体表面的气体可视为二维流体,如能构建二维流体的热力学模型则可深层次分析固体表面的吸附行为。针对二维流体,朱雨航等[21]建立了二维硬碟链流体的分子热力学模型。在此基础上,为了得到适用于实际流体的模型,建立了二维变阱宽方阱链流体的分子热力学模型(SWCF−VR−2D)[22],模型能满意计算单一气体在固体界面上的吸附等温线。本文是这一工作的继续,目的是构建二维流体混合物的分子热力学模型,并考察模型在气体混合物吸附等温线的应用效果。
1 分子热力学模型
1.1 二维变阱宽方阱链流体分子热力学模型
设二维混合流体由K 种分子构成,分别为N1、N2,…,Nk,分子由硬碟组成,链节数为r,硬碟直径为σi,硬碟间具有方阱作用,方阱阱宽和阱深分别为λ2D,i和ε2D,i。则混合流体的亥氏函数表达式可以写为

式中,A为Helmholtz自由能,J;T为温度,K;k为Boltzmann 常数,J/K。上角标“ideal”表示理想项,“mono”为硬碟单体项,“chain”为硬碟成链项。下角标“2D,mix”表示二维混合流体。
理想贡献项表达式与三维流体[23]类似

式中,xi为二维流体中组分i 的摩尔分数;ρ2D,i为组分i 的分子密度;Λi为组分i 的de Broglie 热波长,m,计算式为
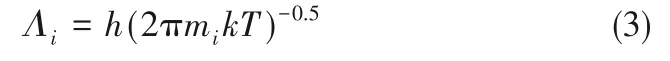
式中,h为Planck常数,J·s;mi为组分i的分子质量,kg。
单体贡献项采用式(4)计算

式中,amono为方阱单体亥氏函数,由硬碟单体排斥作用和硬碟间方阱作用两部分组成


式中,B3为第三维里系数,B3= 16/3 −4 3 /π。w采用式(7)计算

ξ 为二维混合物流体的对比链节密度,计算式为

对于硬碟间方阱作用表达式,式(5)中右边第二项为[20]
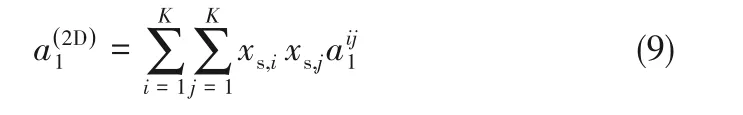
式中,xs,i为组分i的链节分数,表达式为

其中


式中,下角标“ij”表示组分i 和组分j 相互作用,交叉参数采用下列混合规则

式(5)中右边第三项可用式(19)表示

式中,KHD为混合流体的等温压缩系数,表达式为
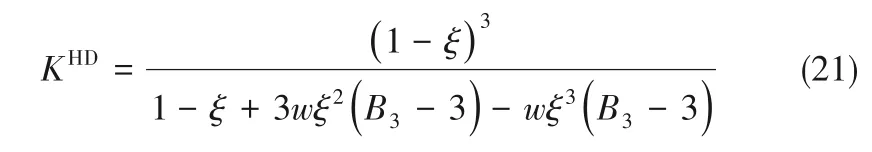
成链贡献项由相邻硬碟成链和相间硬碟成链贡献两部分组成,表达式为

1.2 界面吸附量
当气体被吸附于固体壁面时,气体可视作单分子层吸附,但受到壁面的影响[20],吸附层的亥氏函数应该改写为

式中,εw,i和λw,i分别为壁面与组分i 分子间方阱作用阱深和阱宽。
当混合气体在吸附剂表面达到吸附平衡时,组分i 的吸附层化学势μads,i应与体相化学势μ3D,i相等,即

化学势的值可通过热力学公式导出

式中,三维流体亥氏函数表达式A3D来源于SWCF−VR 模型[23]。当体相温度、压力和组成确定后,组分i 在体相中的化学势μ3D,i也随之确定。而μads,i表达式中有两个未知变量:组分i 的分子密度ρ2D,i和摩尔分数xi。求解式(24)可以得到ρ2D,i和xi,进而得到组分i的界面吸附量

式 中,S*为 吸 附 剂 比 表 面 积,m2/g;NAV为Avogadro常数,mol−1。
2 结果和讨论
2.1 模型参数的确定
模型需要确定6 个参数,分别为r、σ、λ2D、ε2D、λw和εw。σ 和r 为分子结构参数,应与三维流体一致,这里采用SWCF−VR 模型计算结果[23,25−27]。表1 给出了本文涉及到的三维流体的模型参数,其中,λ3D和ε3D分别为三维流体方阱作用阱深和阱宽。
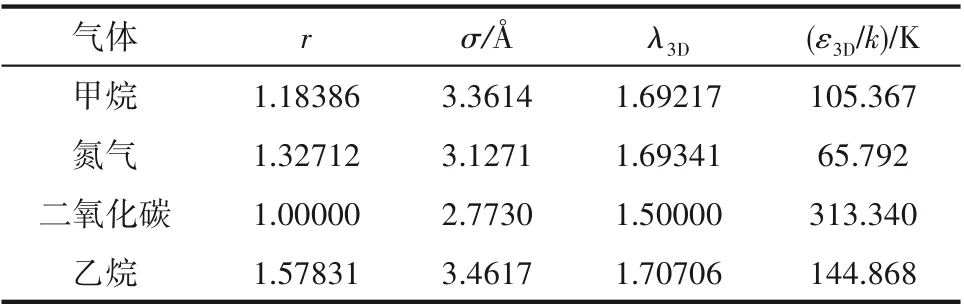
表1 模型参数Table 1 Model parameters
根据经验[17,22,28],本文取λw=0.8165、ε2D/ε3D=0.8。余下的参数λ2D和εw可利用模型关联单一气体吸附数据得到,由于εw表示气体与固体壁面间作用力大小[22],所以其值会随着温度和固体壁面的不同而不同。界面吸附量的模型计算值与实验值之间的平均偏差AAD(%)采用式(28)计算
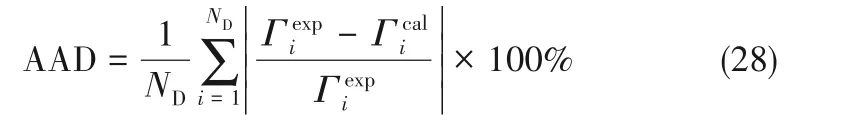
式中,ND为数据个数;和分别为界面吸附量模型计算值和实验值,mmol·g−1。
表2中给出了单一气体的模型关联结果以及所获得的模型参数。
2.2 气体混合物的吸附等温线
采用单一气体吸附数据拟合获得模型参数后,则可直接用SWCF−VR−2D 模型预测气体混合物在固体界面的吸附量,但预测结果偏差较大。实际上,其他如SAFT−VR−2D 模型直接应用于混合物时的效果也并不理想[20]。为提高SAFT−VR−2D模型的计算精度,Castro等[35]在界面吸附量的表达式中添加了两个参数来效验结果,并认为不同流体的吸附面积不一样。前已述及,εw,i体现了组分i 与壁面间作用大小[22],在混合物中,气体分子吸附于界面后将改变界面的性质,从而影响界面与其他分子的相互作用,即单一气体环境下的εw,i与混合物环境下的εw,i将不一样。为此,本文通过引入可调系数fi来体现气体混合物环境下的气体与固体壁面间相互作用能量参数的变化,则式(23)可变为
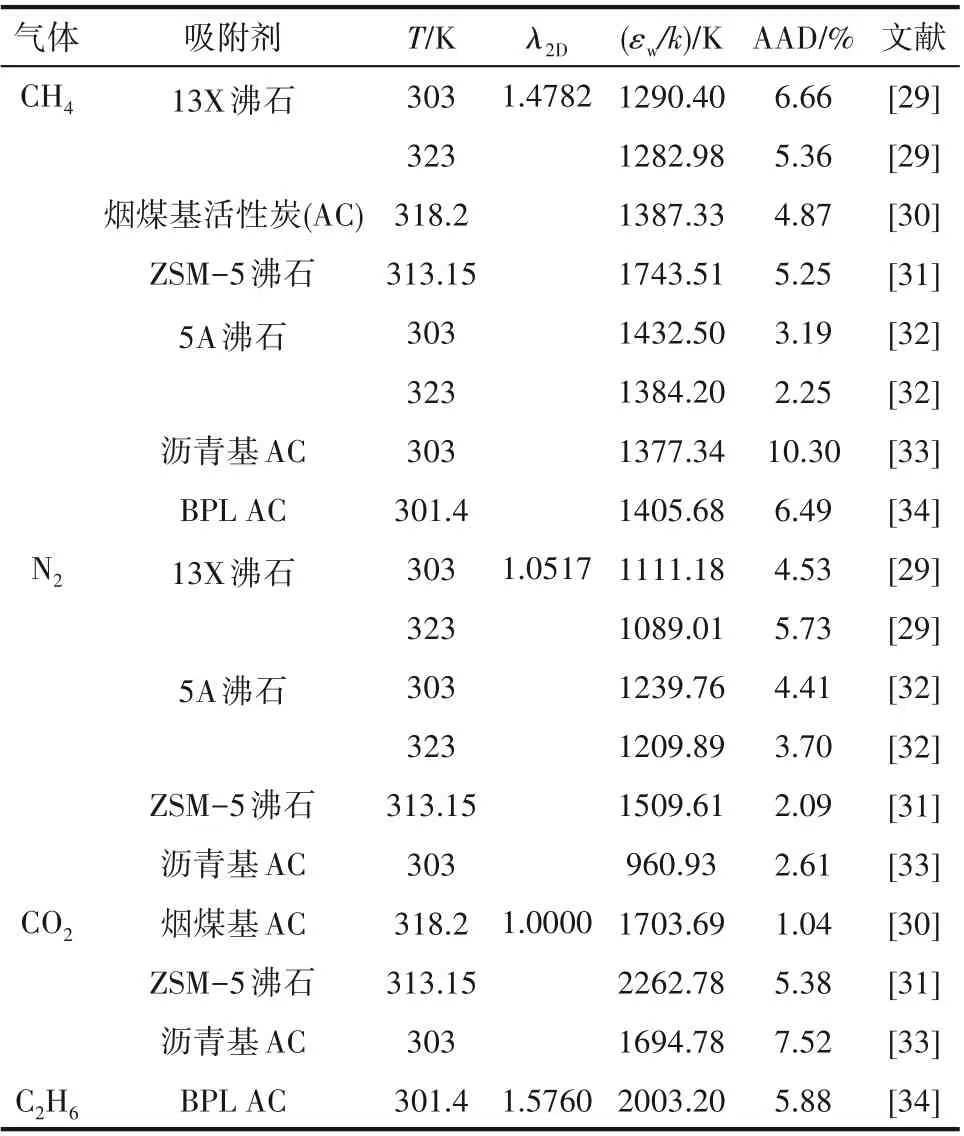
表2 纯物质的模型计算结果Table 2 Calculated results of pure substance

可调系数fi可通过关联混合气体吸附的实验数据得到:利用程序不断迭代得到吸附量的计算值,当计算值与实验值之间偏差最小时,取此时的fi值。相比于单一气体,当fi大于1时,组分i与壁面间作用力增强。当fi小于1时,组分i与壁面间作用力减弱。如吸附剂处于单一气体环境,则fi等于1。另外,组分对界面的影响毕竟有限,fi一般在1附近取值。

图1 甲烷/氮气在13X沸石上吸附等温线的模型计算值与实验值Fig.1 Calculated and experimental adsorption isotherm for CH4/N2 on 13X zeolite
图1 给出了在323 K 下,甲烷/氮气吸附于13X沸石时吸附等温线的实验值[29]和模型计算值。其中,实线为SWCF−VR−2D 模型计算值,点为文献[29]实验值(后图中,线为计算值,点为文献实验值)。由图1 可知,在整个压力范围内SWCF−VR−2D 均能满意计算不同组成下的甲烷和氮气的吸附等温线,平均偏差分别为2.64%和2.63%。如直接利用SWCF−VR−2D 预测(即f1=f2=1),平均偏差分别为7.06%和10.75%。表3 给 出 了IAST[12]、W−VST[13−15]、Flory−Huggins 空穴溶液模型(FH−VST)[36]、PR 2D−EOS[16]以及本模型的计算结果。由表3可知,引入可调系数fi后,SWCF−VR−2D 模型能很好地应用于二元混合气体吸附等温线的计算中,且模型的计算效果均优于其他4种模型。
图2 示意了甲烷/氮气在ZSM−5 沸石上的吸附量随体相组成的变化关系,实验值是在303 K、0.101325 MPa 条件下获得的,直接取自文献[31]。发现SWCF−VR−2D 预测甲烷和氮气吸附量的平均偏差分别为8.91%和15.72%。但引入可调系数后,平均偏差分别降至1.33%和2.41%,且SWCF−VR−2D模型能满意再现体相组成对吸附量的影响。如采用IAST、FH−VST 和扩展的朗缪尔模型(EL−B)[37],发现效果最好的FH−VST 模型对甲烷和氮气的平均偏差也分别为1.66%和10.2%,这说明,SWCF−VR−2D的关联结果要优于上述模型。3.37%。研究还发现,当体相中二氧化碳占比较大(60%~80%)时,随着压力的增加,模型均会低估二氧化碳的吸附量。可能的原因在于二氧化碳与壁面间相互作用(εw/k=1703.69 K)远比甲烷与壁面间相互作用(εw/k=1387.33 K)强,二氧化碳会优先竞争吸附于固体界面。而且二氧化碳分子间的相互作用也较强(ε3D/k=313.34 K),它们在界面上可发生多层吸附,这种多层吸附会随压力升高而进一步强化。本文的模型基础是单分子层的二维流体[22],因而模型将会低估高压下的吸附量。

表3 不同模型对甲烷/氮气吸附等温线计算结果的比较Table 3 Comparison of calculated results by different models for CH4/N2 adsorption isotherm

图2 甲烷/氮气在ZSM−5沸石上吸附量的计算值与实验值Fig.2 Calculated and experimental adsorption amount for CH4/N2 on ZSM−5 zeolite

图3 甲烷/二氧化碳在ZSM−5沸石上吸附量的计算值与实验值Fig.3 Calculated and experimental adsorption amount for CH4/CO2 on ZSM−5 zeolite

图4 甲烷/二氧化碳在烟煤基活性炭上吸附等温线的计算值与实验值Fig.4 Calculated and experimental adsorption isotherm for CH4/CO2 on bituminous−coal−based AC
图5示意了301.4 K时,甲烷/乙烷在BPL活性炭上吸附等温线的实验值[34]与计算值,其中甲烷和乙烷的计算平均偏差分别为4.53%和4.64%。如直接利用模型预测,平均偏差分别为62.88%和8.66%。这也再次说明,引入可调系数fi后,SWCF−VR−2D模型能满意关联烃类混合物的吸附等温线。此外,由图5 还发现,计算偏差随着压力的增大而增大。主要是压力低时,吸附为单分子层吸附,这与二维流体模型建立的假设高度吻合[22],因而计算偏差小。

图5 甲烷/乙烷在BPL活性炭上吸附等温线的计算值与实验值Fig.5 Calculated and experimental adsorption isotherm for CH4/C2H6 on BPL AC
除上述图示结果外,本文还对甲烷/氮气/5A 沸石体系[32]、甲烷/氮气/沥青基活性炭体系[33]、甲烷/二氧化碳/沥青基活性炭体系[33]采用SWCF−VR−2D 计算了吸附等温线,所得结果一并列入表4中,表中也给出了可调系数以及模型的预测结果。由表4 可知,模型的预测结果总的平均偏差为17.62%,引入系数后的平均偏差为5.06%。
应该指出,混合气体中各组分的相互作用、各自与壁面间的作用对吸附的影响机理十分复杂[38],但本文并没有考虑具体的作用力影响,而是将这种影响用εw,i的变化来表示,并通过引入可调系数fi来体现。如果模型的预测效果越差,则拟合得到的fi偏离1 的程度也越大。如二氧化碳/甲烷在烟煤基活性炭上的吸附,模型预测的二氧化碳吸附量的平均偏差高达53.3%,此时,只有将f1调至0.8816 才能得到5.55%的计算偏差;甲烷/氮气吸附于13X 沸石时,拟合所得的f1为1.0003,如取f1=1时,模型预测甲烷的计算偏差也仅为3.54%。这也能说明,fi一定程度上表示混合气体吸附等温线相比于单一气体吸附等温线的偏离程度,εw,i的较小变化会对计算结果造成较大影响。
3 结 论
将SWCF−VR−2D模型扩展至混合流体,并通过引入可调系数以反映界面与气体分子间相互作用能量参数在混合气体中的变化。引入可调系数后,模型能很好地计算甲烷/二氧化碳、甲烷/氮气和甲烷/乙烷等混合气体在不同组成下的吸附等温线,能再现吸附量随组成的变化规律,总的平均偏差为5.06%。
符 号 说 明
A——Helmholtz自由能,J
fi——可调系数
h——Planck常数,6.626×10−34J·s
k——Boltzmann常数,1.381×10−23J/K
N——分子数
NAV——Avogadro常数,6.022×1023/mol
p——压强,MPa
T——温度,K
x——摩尔分数
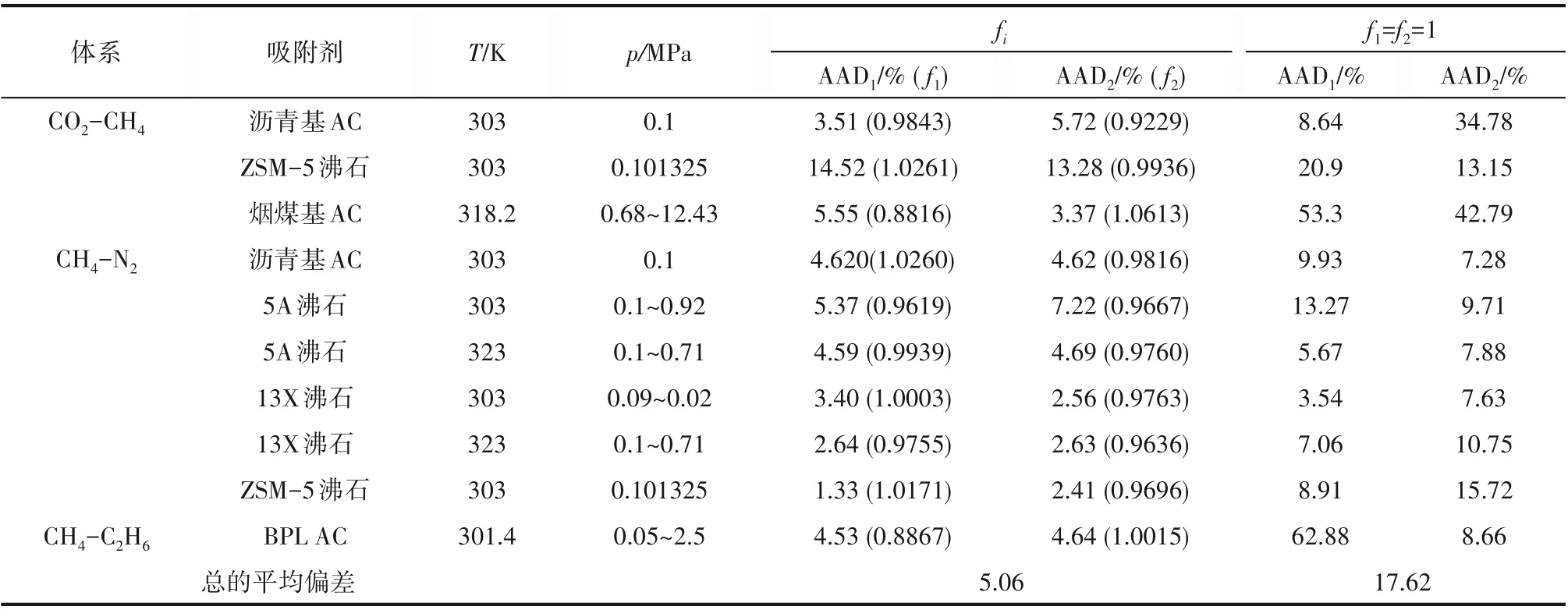
表4 混合气体SWCF-VR-2D的计算结果Table 4 Calculated results of gas mixture by SWCF-VR-2D
μ——化学势,J/mol
ρ——密度,1/m3
上角标
cal——模型计算值
exp——实验值
下角标
ads,mix——吸附层
HD——硬碟间排斥作用项
i——组分
j,j+1——相邻链节对
j,j+2——相间链节对
S——面积
T——温度
V——体积
w——固体壁面
2D——二维流体
3D——二维流体
猜你喜欢
北京航空航天大学学报(2022年7期)2022-08-06
军民两用技术与产品(2021年10期)2021-03-16
哈尔滨工业大学学报(2020年1期)2020-12-21
水上消防(2020年1期)2020-07-24
疯狂英语·新读写(2018年3期)2018-11-29
北京航空航天大学学报(2017年5期)2017-11-23
北京航空航天大学学报(2016年6期)2016-11-16
太空探索(2016年5期)2016-07-12
中学政史地·教学指导版(2014年10期)2015-02-02
物联网技术(2014年3期)2014-04-04
