
健康与腹泻仔猪源A型产气荚膜梭菌的多位点序列分型
2021-03-10 02:41王婧祺戴益民韩春生吴允正王林康张锦华
中国兽医学报 2021年1期
王婧祺,戴益民,王 娇,韩春生,曾 秀,吴允正,王林康,张锦华
(江西农业大学 动物科学技术学院,江西 南昌 330045)
产气荚膜梭菌(Clostridiumperfringens,Cp)既是普遍存在于自然环境中的细菌,又是人类食物中毒以及动物胃肠道疾病、坏死性肠炎和肠出血症的主要病原[1-2]。每年因Cp引发的疾病都会给养殖户造成较大的经济损失[3]。Cp通常根据其产生的几种主要毒素(α,β,ε、ι、NetB和CPE毒素)被分为7种类型(A~G型)[4-5], A型Cp(CpA)是肠道中常见菌群,但在一定条件下也因产生毒素致病,CpA导致的食物中毒也是人类常见的食源性疾病[6]。近年来常有经动物消化道感染CpA,诱发出血性或坏死性肠炎和肠毒血症的报道[4-13]。
随着分子生物学和基因组学的发展,各种分子分型技术被广泛应用于评估细菌种群多样性和Cp的暴发性研究[8-9]。许多研究表明,基于序列分析的多位点序列分型技术(multilocus sequence typing,MLST)有助于了解分离株的种群结构和遗传进化,实验室之间无需交换菌株,通过互联网就可以实现世界范围的流行病学调查[10-11]。
本研究基于Cp公开可用的8个管家基因,使用MLST方法对24株CpA的 ST型进行分析,该24株CpA来源于江西省4个地区的健康和腹泻仔猪肠道。由于Cp为条件致病菌,在健康仔猪肠道同样存在。比较同一地区健康与腹泻来源CpA的等位基因序列的多态性,探究CpA对仔猪致病性的分子机制;分析不同地区健康源CpA和腹泻来源CpA的差异,了解菌株基因序列中的潜在突变和内在联系,以及环境因素对CpA致病性的影响,为制定合理的防控措施提供理论依据。
1 材料与方法
1.1 CpA DNA的制备从实验室保存的菌株中选取24株CpA,菌株来源参见周姣等[12]方法:使用无菌棉拭子从江西省4个地区(南昌、吉安、九江和宜春)的养猪场分别收集7日龄内新生健康和腹泻仔猪的粪便样本(本试验所指腹泻仔猪粪便为常规性粪便不成形或者水样便),按区域进行标记后将收集到的样品立即送往实验室进行菌株分离签定、16S rDNA测序和多重PCR毒素分型。对选取的24株CpA使用磁珠法DNA提取试剂盒(Cat No:DP712,TIANGEN,China)提取DNA,使用NanoDrop 2000(Thermo Electron Corporation,USA)在260和280 nm的波长下测定DNA浓度。
1.2 MLST方案本研究MLST的管家基因方案参照文献[14],除7个常见的管家基因外,还包括编码α毒素的plc基因。表1列出了选用管家基因的序列长度以及相应引物和蛋白产物。
1.3 管家基因序列扩增24份DNA样品由8个管家基因引物进行PCR扩增。25 μL PCR扩增体系包括:2 μL模板DNA,1×含有Mg2+的PCR缓冲液,0.2 mmol/L脱氧鸟苷三磷酸盐混合物,2.5 U Taq DNA聚合酶和每种引物800 nmol/L,剩余体积由蒸馏水补齐[15]。PCR扩增条件参照NAKANO等[16]的研究,通过水平琼脂糖凝胶电泳检验所有PCR产物。将PCR扩增片段连接T载体后转入感受态细胞,筛选阳性克隆后送至上海生工生物工程有限公司进行双向测序。
1.4 核苷酸序列登录号本研究中发现的每个独特序列上传至GenBank数据库,获得登录号如下:dutMK626534-MK626541,recAMK729665-MK-729729675,tpiMK729676-MK729682,sodMK729-683-MK729689,ddlAMK729690-MK729699,gmkMK729700-MK729710,glpKMK729711-MK729-715,plcMK729716-MK729726。

表1 管家基因的选择和引物序列(参考菌株:ATCC 13124)
1.5 等位基因ST型编号和多样性分析每个管家基因的所有等位基因按测序获得等位基因编号。产气荚膜梭菌ATCC13124(登录号NC_008261.1)的核苷酸序列从NCBI数据库获得并用于后续的比较分析。修剪每个等位基因的序列长度,其长度范围从429 bp(ddlA)至574 bp(glpK)。8个管家基因的每个等位基因按一定顺序排列形成等位基因谱,并基于独特的等位基因谱生成ST型给予序列号。多个ST型由BURST方法聚集成不同的克隆复合物(cloning complex,CC),克隆复合物是由存在部分相同编号的等位基因的多个ST组成,并且每个克隆复合物被任意分配一个数字编号。使用START 2软件(https://pubmlst.org/software/analysis/start2/)构建ST型系统发育树并用统计学方法分析突变率[17]。根据2个ST型之间的距离(距离代表遗传变化的程度)评估各分支的关系。如果聚集在同一亚组中是同一节点的分支,距离近,表明遗传相似度高。其他数据分析,包括对健康源和腹泻源CpA的聚类分析,则使用MAGE 7.0软件 (http://www.megasoftware.net/)完成。
2 结果
2.1 CpA菌株的获得从通过16S rDNA测序和多重PCR分型被鉴定为CpA的菌株中选取24株,包含健康源和腹泻源各12株,样品覆盖江西4个地区,每个地区3株。
2.2 基因多样性分析MLST管家基因的多样性显示在表2中。对比MLST方案的多位点连锁不平衡标准指数(standardized)和古典指数(classical),2种指数的观察方差都大于相对应的最大方差值(表3)。本试验MLST方案中管家基因长度在429 bp(ddlA)和574 bp(glpK)之间,plc、recA和gmk这3个管家基因具有更高的突变。无论是否添加α-毒素调控基因(plc),整体ST数量都没有变化,并且除去plc基因进行聚类分析可观察到的克隆复合物聚集没有明显变化。
每个管家基因存在数量不等的等位基因,数量在4(glpK)到10(plc)之间,等位基因中存在的可变位点的比例为1.8%(tpi)至6.1%(plc)。8个管家基因的等位基因同义突变与非同义突变的比率范围是dN / dS = 0.057 0(sod)至dN / dS = 0.360 9(recA),计算比率都小于1。在12株健康源CpA中,单个管家基因的等位基因数不超过5;但腹泻源CpA中管家基因存在更多的突变,单个管家基因突变数更高。腹泻源CpA的plc管家基因具有更高的多态性,存在9种等位基因,而健康源CpA中仅存在以plc-ST3为主的4种等位基因。在整体分析中,glpk等位基因数量最少,Sawyer游程检验中MCF的P值为0.517,MUF的P值为0.515,这也与其他管家基因不同。
2.3 健康源与腹泻源CpA的聚类分析选取的24株CpA产生24个ST型(图1)2个克隆复合体,CC1是其中较大的一组,其余的ST型是独立的。CC1是以ST-1为中心的克隆复合体,其中聚集了75%的健康源CpA,江西吉安和宜春地区的部分腹泻源ST型与南昌、九江地区健康源ST型的遗传距离相对接近并聚集在同一亚组。相比之下,CC2是来自2个地区样本明确定义的复合体,含有4种腹泻源CpA。通过最小生成树的聚类分析,不同地区的CpA菌株之间的亲缘关系并没有表现一个地区的菌株呈现单独的聚集。
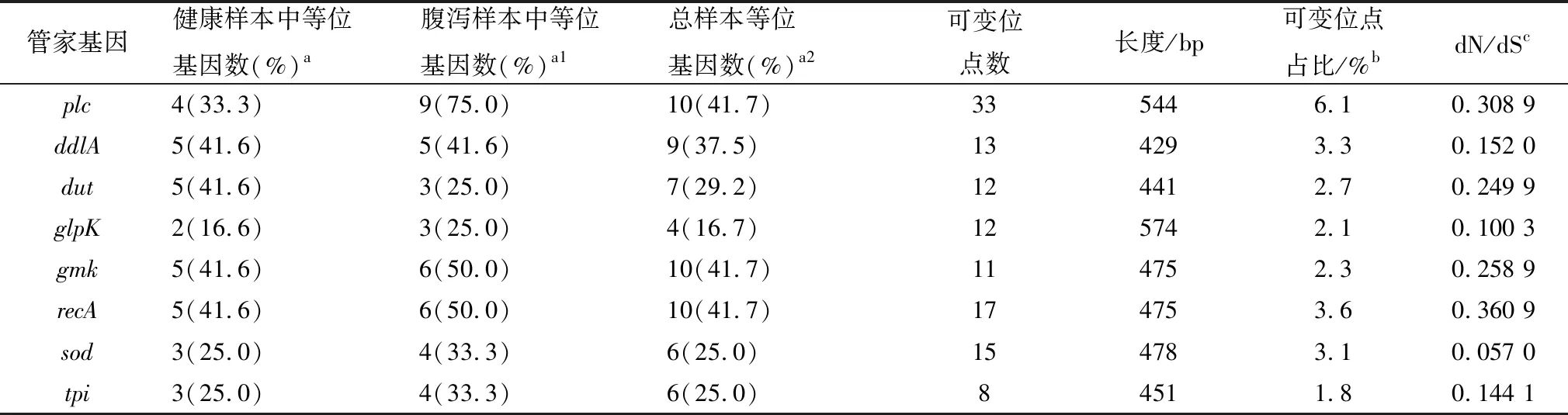
表2 CpA管家基因的遗传多样性
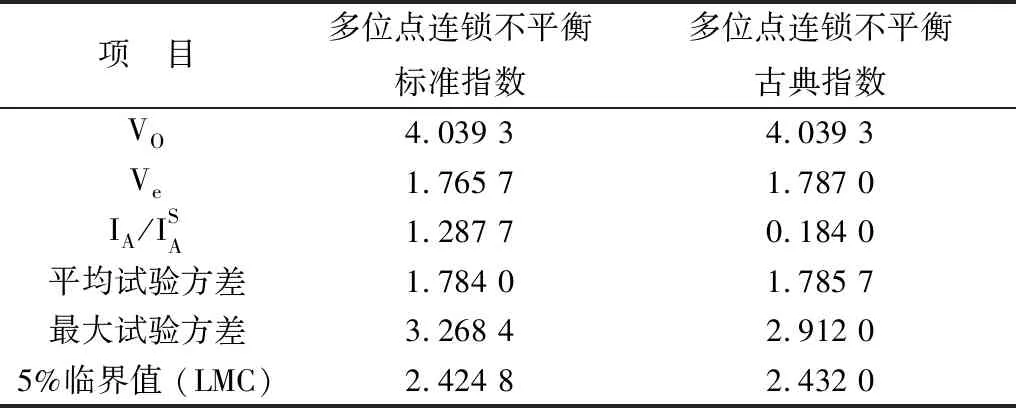
表3 多位点连锁不平衡检测
2.4 不同地域的健康源和腹泻源CpA聚类分析对健康源和腹泻源CpA分别聚类分析(图2),不同地区的健康源CpA均存在较高的相似性,地域差异对健康源CpA影响较小。除少数菌株外,腹泻源CpA中同一区域菌株的聚集程度明显,腹泻源CpA的地理特异性相对较高。江西九江地区的2个ST(JJST22、JJST23)是同一节点的2个分支,有高度相关性,但这2个ST与其他地区的腹泻源ST相比,相似性较低,差异显著。

系统发育树参考株为ATCC13124,已在图上标记;绿色为健康样本分离株,红色为腹泻样品分离株
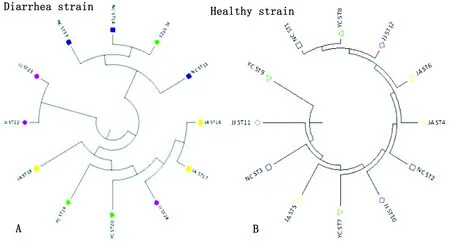
图2 腹泻源与健康源CpA菌株的聚类分析(MEGA软件)
以区域分界对健康源和腹泻源CpA聚类情况的分析(图3),江西南昌和九江地区的健康源和腹泻源CpA分别聚类成2个亚组,健康和腹泻区分明显;南昌的腹泻源CpA是源自同一节点的分支,有高亲缘性如图3A,但有些地区呈交叉分布。
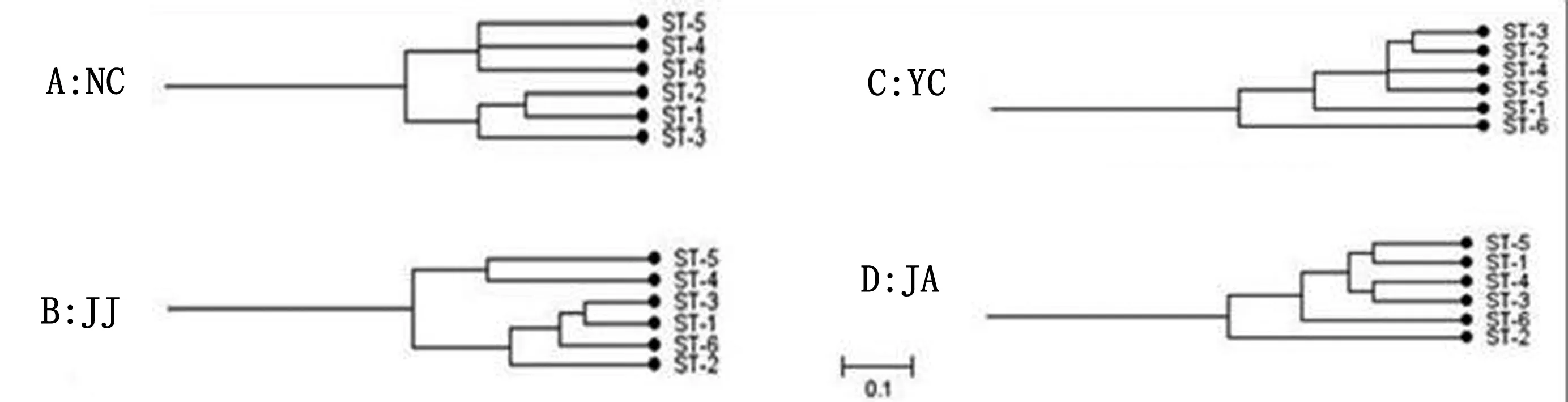
A:NC.南昌;B:JJ.九江;C:YC.宜春;D:JA.吉安。健康样本分离株 ST1-3;腹泻样本分离株 ST4-6
3 讨论
多位点连锁不平衡标准指数(standardized)和古典指数(classical)表明本试验的MLST方案存在明显的连锁不平衡,说明8个管家基因的等位基因之间是具有一定程度的遗传突变关联性,MLST分析结果具有较高的可信度。等位基因的dN/dS值远小于1,表明这些管家基因不受外部环境选择压力的影响。等位基因的数量在一定程度上反映了管家基因的多样性。在腹泻源CpA中管家基因plc、gmk和recA的等位基因数量均高于健康组,说明腹泻源CpA中管家基因的多样性较高。
所有类型的Cp都能产生α毒素。许多研究已经证明了α毒素对Cp病的影响[18]。因此,本试验在Cp常见的7个管家基因的基础上,添加plc基因作为管家基因来评估α毒素基因在核苷酸水平上的突变情况。EFFAT等[19]研究发现在10株不同毒素类型Cp中,α毒素基因平均有 1.3%的核苷酸序列不同,有1.2% 的氨基酸序列不同,但不同菌株产生的α毒素仍具有相同的生物学功能。本研究结果表明同一毒素类型的CpA,其α毒素(plc)基因具有较高的突变,突变位点多达33个(33/544,6.1%核苷酸序列不同)。腹泻源与健康源CpA间存在的等位基因相似,但数量不同,腹泻源CpA中plc等位基因数量明显高于健康源,呈现较高的多样性,最大卡方检验表明该结果具有统计学意义。但无论是否添加α-毒素调节基因(plc)作为管家基因,ST型整体数量都是不变的,聚类情况差异不明显。
本研究在不同地区健康和腹泻仔猪粪便分离到的24株CpA,根据MLST方案得到24个ST型,呈现较高遗传多态性。该24株CpA覆盖江西省的4个主要行政区,在地理区域的选择上有一定的代表性。24个CpA均来自相同宿主仔猪,确保宿主因素不影响菌株聚类分析。有报道称同种宿主来源的菌株所在的环境区域不同,因每个区域都有自己独特的自然选择突变而呈现多种ST型;当同种宿主来源的Cp承受同一地区的选择压力时,可以发现相同地区的菌株也具有不同的ST型[2,20]。ENGSTRÖM等[21]在对7个农场中不同健康程度的鸡群分离到的Cp表型和基因型的研究中,也发现来自同一地区、同一宿主不同健康程度样本分离到的Cp表现出多种ST型。
健康源和腹泻源CpA遗传进化树分析表明:腹泻源和健康源的CpA分别相对聚集,CC1是以ST-1为中心的克隆复合体,聚集了75%的健康源CpA,还有部分腹泻源CpA零星分布于健康源CpA聚集的CC1亚组中,总体来说不同地区健康源CpA具有一定的亲缘关系。而江西吉安地区腹泻源CpA(ST16、ST18)与其他地区健康源CpA(ST2、ST12)有较高的相似性,该地区的健康源与腹泻源CpA呈现交叉分布的特点。
从UPGMA方法构建遗传进化树分析,腹泻源CpA的地理特异性相对较高。CC2亚组中同一地区的腹泻源CpA聚集程度明显,如ST13、ST14和ST15,这3株均分离自南昌,因此这些菌株可能具有相同的遗传背景。南昌的3株CpA和宜春的1株(ST21)源自同一分支,存在明显的相关性,可能由于在江西省的地理分布图中,南昌和宜春是相互接壤的,菌株在基因突变过程中受到横向水平的影响。同一地区健康源和腹泻源菌株的聚类分析,大部分地区健康源和腹泻源CpA聚类明显,江西九江地区的健康源和腹泻源CpA呈交叉分布,较近的亲缘关系提示我们该地区健康源CpA具有潜在致病的可能。
本研究第1次利用MLST技术分析从江西省4个地区分离到的健康和腹泻仔猪源CpA。聚类分析结果表明CpA具有较高水平的遗传多态性,在基因水平上管家基因glpk相对保守,recA和plc基因变异程度较大,增加或者减去plc基因,对整体聚类情况无明显影响。健康源和腹泻源CpA成2个亚组聚集,CC1是以ST-1为中心的克隆复合体,聚集了75%的健康源CpA。此外,腹泻源CpA有一定的地理亲缘性,其可遗传的突变有一定的规律和方向性,因此,推测CpA在仔猪腹泻中的影响在一定程度上与地域环境相关。因本试验样本相对较少,有待进一步的试验结果验证。
猜你喜欢
环球时报(2022-09-20)2022-09-20
考试与评价·高二版(2021年3期)2021-09-10
智慧健康(2021年17期)2021-07-30
国际检验医学杂志(2021年7期)2021-04-15
今日农业(2020年24期)2020-12-15
数学物理学报(2020年5期)2020-11-26
天然产物研究与开发(2018年8期)2018-09-10
现代检验医学杂志(2016年5期)2016-08-20
公民与法治(2016年14期)2016-05-17
兽医导刊(2016年12期)2016-05-17
