
单空缺石墨烯负载的Pd单原子催化剂上NO还原的密度泛函理论研究
2021-04-09 06:49张芳芳韩敏赵娟凌丽霞章日光王宝俊
化工学报 2021年3期
张芳芳,韩敏,赵娟,凌丽霞,章日光,王宝俊
(1 太原理工大学化学化工学院,山西太原030024; 2 太原理工大学煤科学与技术教育部和山西省重点实验室,
山西太原030024)
引 言
随着社会的发展,人们对机动车的需求不断增加,私家车数量逐年快速递增,机动车的尾气污染已成为环境面临的重要挑战[1]。汽车尾气含有大量CO、NOx、碳氢化合物、SO2以及粉尘等,有害气体严重超标。其中NO 的毒性强,长期暴露在NO 限定值外的环境中,容易诱发各种疾病,严重影响人体的健康。另外,汽车尾气中的碳氢化合物与NOx在太阳的作用下,与大气中的VOC(挥发性有机化合物)发生光化学反应会造成二次污染,形成以臭氧和过氧乙酰硝酸酯(PAN)等细颗粒物为主的烟雾污染物,即光化学烟雾[2]。此外NOx还容易与氧气发生反应形成硝酸,造成酸雨,酸雨容易损伤农作物、森林、土壤等。除了影响人类身体健康,造成酸雨、光化学烟雾外,NOx还会引发臭氧层空洞、全球气候变暖等问题[3],因此脱除NOx成为重要的环保课题。氮氧化合物中NO 占95%以上,因此对NO 的还原显得至关重要。
NO 还原的催化剂包括贵金属[4-5]、金属氧化物[6-8]和分子筛[9-11]等。贵金属催化剂,特别是Rh、Pt和Pd,作为三元催化剂,可以还原NO 以及氧化CO和未燃烧的碳氢化合物,成功地吸引了广泛的关注和研究[12]。Loffreda 等[13]通过研究Pd、Pt、Rh 三种金属不同暴露面上NO 的吸附,发现Rh 的绝大多数暴露面和Pd的个别暴露面对NO的吸附和解离效果较好。与Rh、Pt 催化剂相比,Pd 基催化剂具有良好的热力学稳定性、低温下的高活性。Pd 物质能够在低温下化学吸附NO以在冷启动期间减少NOx排放,因此有大量的实验研究致力于探索Pd 基催化剂上H2选择性还原NO。与Pd 块相比,Pd 纳米团簇具有不同的结构和电子性质,这使其对催化剂的制备具有极大的吸引力[14]。长期以来,减少贵金属的用量,同时保持催化反应活性一直是重要的研究课题[15]。单原子催化(SAC)可以在均相和非均相催化之间架起桥梁,最大限度地利用原子。单原子催化的优点是高催化活性、高选择性和高利用率。在微观层面上,单个原子的特征(例如低配位环境、量子尺寸效应和金属-载体相互作用)使单原子催化具有优异的催化活性、选择性和稳定性[16]。将单原子Pd 负载到不同载体上催化NO 还原已被众多实验研究过,Paredis 等[17]对ZrO2载体负载Pd 纳米颗粒催化剂上H2还原NO 的过程进行了研究,结果表明这个催化剂对N2具有极好的选择性。Yin 等[18]通过DFT 方法对MgO 负载的单原子Pd 和Pd4簇上CO 还原NO 的反应机理进行了研究,包括协同反应路径与逐步反应路径,结果表明在Pd/MgO 和Pd4/MgO 上逐步反应路径比协同反应路径容易进行。Pd/CeO2(111)上CO还原NO 的DFT 研究显示单原子Pd的d电子和CeO2氧空缺的相互作用是提高催化性能的关键[19]。
石墨烯是一种由sp2键结合的碳原子以六方晶格排列的平面薄片。由于其独特的电子特性,最近在材料科学和凝聚态物理领域引起了广泛关注[20-21]。石墨烯上碳空位的存在显著影响石墨烯的理化特性和磁性,纳米颗粒负载于缺陷石墨烯比负载于完美石墨烯表现出更强的表面反应性。Jia等[22]对单空缺石墨烯负载Pd 单原子催化剂上的CO氧化反应进行了研究,证实了石墨烯负载的Pd催化剂对CO 氧化有着高的催化活性。在石墨烯负载的单原子催化剂上进行NO 的还原,之前已经有过很多实验进行论证。在单空缺石墨烯负载的单原子Si催化剂(Si/SVG)上,NO 经过二聚体构型生成产物N2O[23]。Fe/SVG 上也是通过二聚体路径将NO 还原为N2O,接着N2O 分解为N2[24]。在Al 和Si 沉积的氧化石墨烯(GO)上进行了NO 的还原,研究表明在Al/Si/GO 上NO 的二聚体吸附能大于单个NO 分子,二聚体路径易于直接解离路径,其中Si/GO 的催化活性大于Al/GO[25]。单空缺石墨烯负载的单原子Ti 催化剂被用于CO2催化加氢制甲酸反应,表明此催化剂具有高的催化活性[26]。此外石墨烯上含氧官能团的存在对催化剂的催化性能有重要的影响[27-28]。但在实验中可以通过热退火以及化学还原等方法将氧化石墨烯上的含氧官能团还原,得到还原性的石墨烯[29-30],使得表面的π 电子恢复[31]。Erickson 等[32]用透射电子显微镜(TEM)对合成的还原氧化石墨烯结构进行表征,得到还原石墨烯的微观结构为不含任何含氧官能团的单层石墨烯。
NOx的选择性催化还原(SCR)是调节NOx排放的重要策略之一。在过去的几十年中,NO 与H2、CO、NH3和碳氢化合物的反应已在实际应用中进行了广泛研究[33-34]。在这些还原剂中,H2-SCR 已经引起极大的关注,因为H2易于在废气流中产生,并且其燃烧产物H2O 对环境无害[35]。H2还原NO 的产物为N2和H2O,还原产物清洁无污染,而且H2供给方便、易储存,对于移动源氮氧化物的消除具有重要意义,被认为是最有应用潜力的NO 还原剂。另一方面,H2作为一种活泼的还原剂还原NO,可在低温条件下进行,其催化剂多为贵金属催化剂,这使低温还原NO成为现实。
在本工作中,首先对单空缺石墨烯负载的Pd单原子催化剂上NO 的活化及相关中间体的反应进行了研究,然后探讨了产物N2与NH3的生成路径,阐明了在Pd/SVG 上H2还原NO 的反应机理。最后,得到了Pd/SVG催化剂针对NO还原的活性与选择性。
1 模型构建及计算方法
本研究在Materials Studio 8.0 软件包Dmol3模块上进行。对原始石墨的几何结构进行优化,得到的晶格参数为a=b=2.466 Å(1 Å=1.0×10-10m),其结果和实验值吻合[36]。选取了单层石墨烯,然后在完美石墨烯中去除一个C 原子形成单空缺石墨烯(SVG),再将一个Pd 原子吸附在空缺位上,形成Pd/SVG 结构。在实验上已成功地将Pd 纳米颗粒高分散性地负载在石墨烯上[37],且单空缺石墨烯上负载的单原子Pd、Si、Ni、Fe等催化剂已被广泛地用于CO的氧化[23]、CO 偶联制草酸二甲酯[38]以及Hg0的吸附[39]等研究中。通过对Pd 在超胞分别为p(5×5)、p(6×6)和p(7×7)的SVG 上的形成能进行计算,得出它们的形成能分别为-546.7、-557.5 和-554.1 kJ·mol-1,可见Pd 在p(5×5)的SVG 表面上形成能较小,而在p(6×6)和p(7×7)的SVG 表面上形成能相差不大,且结构很稳定,因而本研究选用p(6×6)的Pd/SVG 对NO 的还原进行研究[37]。真空层设置为15 Å。在计算过程中,由于碳材料结构中存在大量的无定形碳及其他缺陷,使其稳定性远远低于单晶石墨,与石墨片层六元环内的C C 键相比,缺陷及边界处的C—C 单键键能较小,易裂解形成高活性悬空键,因而将边缘C 原子固定来避免石墨烯的悬空效应[20]和边界不饱和效应[40]。优化后的Pd/SVG 的结构图见图1,包括顶视图(左)和侧视图(右),红色的表示固定的C原子。
所有计算采用广义梯度近似(general gradient approximation, GGA),泛函形式为PBE(Perdew-Burke-Emerhof)。采用了双数值加极化基组(double-numeric basis with polarization functions,DNP),对所有小分子采用了全电子(all electron)处理,而所有金属原子采用了有效核电势(effective core potential,ECP)[38]。为了实现快速的电子收敛,热拖尾(smearing)的参数设置为0.005 Ha(1 Ha=2625.5 kJ·mol-1)。受力、能量和位移收敛标准分别是4×10-3Ha/Å、2×10-5Ha和5×10-3Å。其布里渊区k点设置为3×3×1。根据NO 还原实验中检测到的中间体以及相关的理论研究[41-42],拟定反应过程中可能发生的不同路径,需对每一步基元反应过程的反应物和产物进行结构优化。过渡态的搜索采用完全线性同步/二次线性同步(complete LST/QST)方法,先LST优化,再通过共轭梯度最小化和QST的最大化循环计算,最后达到收敛标准。进而对基元反应进行过渡态确认(TS confirmation),确保每步基元反应的过渡态直接连接着此过程的反应物和产物,来确定每个中间态。最后对过渡态进行频率分析,确保其准确性。吸附能Eads、活化能Ea、反应热Er和形成能Ef的定义如式(1)~式(4):
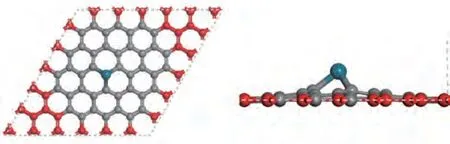
图1 Pd/SVG表面的优化模型(左图和右图分别是顶视图和侧视图)Fig.1 The optimized slab models of Pd/SVG surface(the left and right are top and side views)
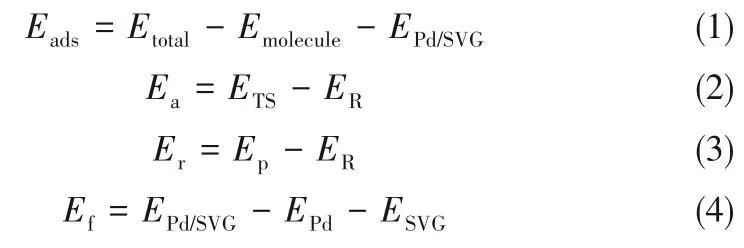
式中,Etotal表示平衡状态时吸附物吸附于表面体系的总能,Emolecule表示吸附物种的能量,EPd、ESVG和EPd/SVG分别是单原子Pd、SVG 和Pd/SVG 的能量。吸附能是衡量吸附物质和催化剂之间相互作用强弱的重要指标,当Eads越负时,吸附物与催化剂表面的作用越强。ER、ETS和EP则分别表示基元反应中反应物、过渡态和产物的能量。Ea越小,表明反应越容易进行。Er为正说明反应吸热,反之为放热反应。
2 结果与讨论
2.1 Pd/SVG上NO的活化
Pd/SVG 上NO 的活化包括NO 的直接解离、NO加氢形成NOH和HNO,这三步的基元反应如下:
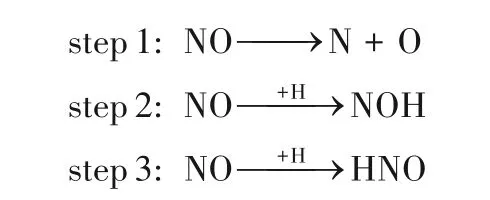
反应的势能图以及对应的反应物、产物、过渡态构型见图2。在Pd/SVG 上NO 的吸附能为-100.2 kJ·mol-1,NO 以N 端稳定吸附在Pd 原子上,N—O 键长由始态的1.191 Å,经过渡态TS1 的2.456 Å 拉长到3.609 Å。直接解离形成N 和O 的能垒高达513.8 kJ·mol-1,可见NO的直接解离很难发生。同样地,在其他催化剂上NO 直接解离的能垒也很高。在Si/SVG 上NO 的直接解离能垒为377.2 kJ·mol-1[43],在Au(111)和Pd(211)上的直接解离能垒分别为381.1和288.4 kJ·mol-1[44-45]。NO 在H 辅 助 作 用 下 有 两 种反应方式,当H 原子攻击NO 的O 原子时,经过渡态TS2 生成NOH,H—O 键长由3.045 Å 经TS2 的1.002 Å 变为0.975 Å,克服的能垒为129.3 kJ·mol-1,比NO的直接解离能垒低。与NO 在Pd(211)和Pd(111)上加氢生成NOH 的能垒相差不大,分别为131.7 和156.3 kJ·mol-1[45-46]。而H 原子攻击NO 的N 原子时,N—H键长由2.385 Å经TS3的1.707 Å变为1.048 Å,生成中间体HNO,克服的能垒仅为67.0 kJ·mol-1,这比在Pd6/TiO2、Pt(111)和Pd(110)上NO 加氢生成HNO所需要的能垒低很多。NO 在Pd6/TiO2上加氢生成HNO 需要克服的能垒为103.0 kJ·mol-1[41],在Pt(111)上需要克服的能垒为97.4 kJ·mol-1[42],在Pd(110)上克服的能垒更高,为158.2 kJ·mol-1[47]。比较在Pd/SVG 上NO 活化的三种形式可见,NO 加氢形成HNO需要克服的能垒最低,是NO 活化的主要方式,其次是NOH 的生成。比Rh(111)上NO 加氢生成HNO(152.4 kJ·mol-1)和NOH(149.6 kJ·mol-1)的能垒都要低[48]。对NO 的活化及中间体HNO 和NH2O 的相关反应的过渡态进行了Mulliken 电荷分析,表1 列出了过渡态结构中吸附物种各原子的Mulliken电荷以及整个吸附物种的电荷。由表1 可知NO 加氢形成HNO 与NOH 过程中,Pd/SVG 转移到吸附物种的电荷数远远小于NO 直接解离过程中转移的电荷数。从这三步基元反应的能垒可知,NO 直接解离所需要的能垒很高,远远大于NO 加氢过程所需要的能垒。表明反应过程中Pd/SVG 向吸附物种转移的电荷越多,过渡态结构中催化剂与吸附物种之间的作用更强,导致反应所需要的活化能越高。

图2 Pd/SVG上NO的活化势能图和对应的反应物、过渡态以及产物的构型Fig.2 Potential energy diagram of NO activation with corresponding configurations of initial states,transition states and final states
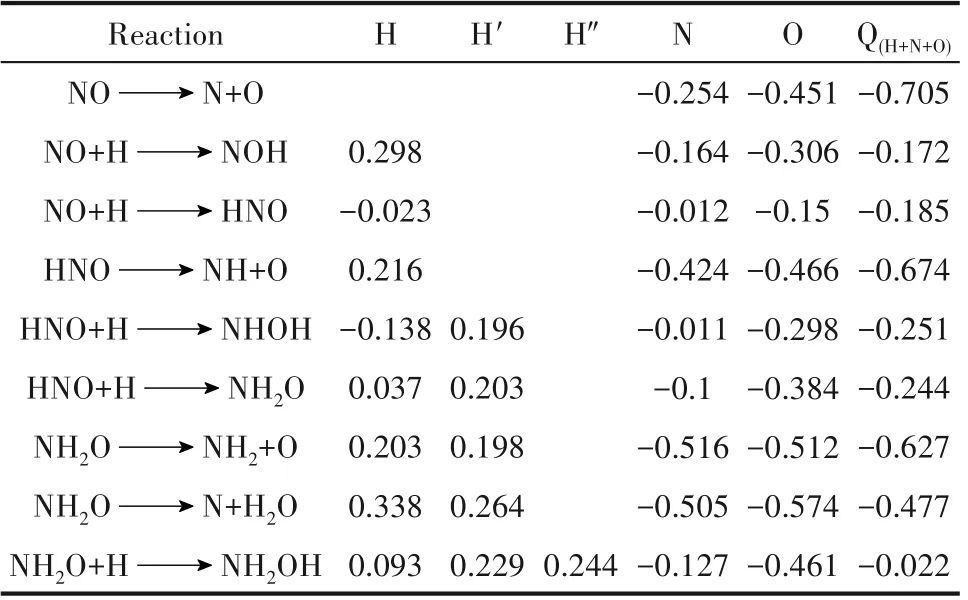
表1 各基元反应过渡态结构中吸附物种各原子的Mulliken电荷以及整个吸附物种的电荷Table 1 The Mulliken charge of each atom of the adsorbed species in the transition state structure of each elementary reaction and the charge of the entire adsorbed species
2.2 H2还原NO过程中相关中间体的反应
NO 最容易通过加氢形成HNO,接下来是HNO基元反应如下:
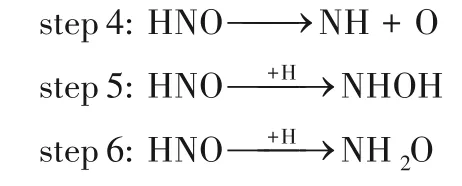
在Pd/SVG 上HNO 直接解离为NH 与O,及HNO加氢生成NHOH 均不易进行,能垒分别为348.8 和213.5 kJ·mol-1。当H 原 子 攻 击HNO 的N 原 子 时,N—H 距离由2.453 Å 缩短为1.027 Å,经过渡态TS6生成NH2O 克服的能垒仅为41.9 kJ·mol-1,反应势能图见图3(a)。这比Pd6/TiO2上HNO 加氢生成NH2O的能垒低,在Pd6/TiO2上这一步的能垒为91.5 kJ·mol-1[41]。由表1 可知,这三个基元反应中HNO 加氢生成NH2O 转移的电荷最少,反应过程的能垒也最低,最易于发生。
HNO 加氢易于生成NH2O,NH2O 的基元反应如下:
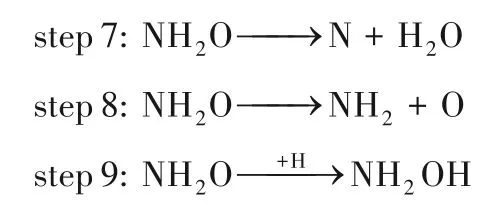
NH2O相关反应的势能图及对应的反应物、过渡态及产物构型见图3(b)。NH2O可经过渡态TS7直接解离为N 与H2O,也可经过渡态TS8 解离为NH2与O,需要活化能分别为332.8 和233.4 kJ·mol-1,可见NH2O 的直接解离很难发生。而在H 的辅助下,当H原子进攻NH2O的O原子经过渡态TS9得到NH2OH,O—H 距离由反应物的2.687 Å 逐渐减小为产物的1.060 Å,克服的能垒仅为86.4 kJ·mol-1。由表1可知NH2O 加氢生成NH2OH 过程中,过渡态结构中催化剂转移到吸附物种的电子数远比NH2O 直接分解反应转移的电子数要少,所以NH2O 易于加氢生成NH2OH。
2.3 产物N2的生成
对Pd/SVG上生成N2的后续基元反应如下:
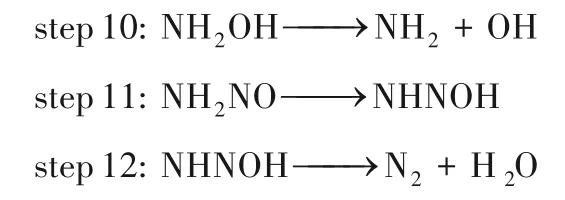
在Pd/SVG 上通过NH2NO 中间体生成N2的路径见图4。首先NO 活化生成HNO,HNO 加氢直到生成NH2OH。NH2OH 解离为NH2与OH,N—O 键长由1.436 Å 变为2.750 Å,NH2与OH 的稳定吸附均在Pd上,此基元反应经过渡态TS10 克服的能垒为82.5 kJ·mol-1。在Pd/SVG 上NH2极易与NO 生成NH2NO,这个过程是自发进行的。在Mn3O4(110)和Mn 掺杂的TiO2催化剂上NH3还原NO 的DFT 研究发现,NH3解离生成的NH2与气态NO 生成NH2NO 也为自发反应[49-50]。然后NH2NO 通过氢转移异构化形成NHNOH,在这个过程中一个N—H 键断裂,H—O 键由反应物的2.373 Å 经过渡态TS11 的1.371 Å 缩短为0.991 Å,新的H—O 键形成,此过程经过渡态TS11,跨越的能垒为144.3 kJ·mol-1。最后NHNOH中的N 上H 转移到O 上,H 与OH 键结合形成H2O。伴随着H2O 的形成,产物N2形成,这一步的活化能为128.5 kJ·mol-1。同样地,在单原子Mn掺杂的TiO2催化剂上,NH2与NO 自发生成的NH2NO 也经分子内氢转移异构化形成NHNOH,然后再解离形成N2和H2O,NH2NO 异构化的过程需要克服的能垒为138.9 kJ·mol-1[50]。可见在Pd/SVG 上经NH2NO 中间体生成N2与H2O的路径与Mn-TiO2上的相似。

图3 Pd/SVG上NO还原的相关中间体解离加氢的势能图和对应的反应物、过渡态以及产物的构型Fig.3 Potential energy diagram of related intermediates in the reduction of NO together with corresponding configurations of initial states,transition states and final states
2.4 产物NH3的生成
生成NH3的后续路径基元反应步骤如下:

Pd/SVG 上生成NH3的路径也为NO 加氢活化生成HNO,然后HNO 加氢生成NH2OH,NH2OH 解离后形成NH2,这些步骤和经NH2NO 中间体形成N2的路径一样。最后NH2加氢经过渡态TS13 生成产物NH3,这个步骤需要克服56.0 kJ·mol-1的能垒。在这个过程中,吸附在C上的H转移到NH2的N上,N—H键的距离由反应物的2.766 Å经过渡态的1.564 Å到NH3的1.023 Å。表面的OH加氢生成H2O,需要的活化能为96.5 kJ·mol-1。从NO活化到生成NH3的过程中,决速步骤为NH2O 加氢生成NH2OH,需要的活化能为86.4 kJ·mol-1。

图4 N2与H2O生成势能图及对应的反应物、过渡态及产物构型Fig.4 Potential energy diagram of correlated reactions of the formation of N2 and H2O together with corresponding configurations of initial states,transition states and final states
2.5 产物N2和NH3的选择性
对于N2与NH3的生成,从NO 活化到中间体NH2的形成,经历了相同的过程:NO 活化生成HNO,HNO 继续加氢生成NH2O 和NH2OH,NH2OH 解离为NH2与OH。然后NH2经过不同的路径导致不同产物的形成:当NH2与NO 结合生成NH2NO 时,产物为N2;当NH2加氢时产物为NH3,路径如图6 所示。生成N2的决速步为NH2NO 分子内氢转移生成NHNOH,所需要的活化能为144.3 kJ·mol-1。NH3生成的决速步为NH2O 加氢生成NH2OH,对应的能垒为86.4 kJ·mol-1。比较生成这两个产物的决速步骤的能垒可见,催化剂Pd/SVG 上NH3的形成更容易。因此,Pd/SVG 上H2还原NO 的产物中NH3的选择性较高。
3 结 论
采用密度泛函理论(DFT)方法对Pd/SVG 上H2还原NO 的反应机理进行了研究,考察了NO 的不同活化方式及可能产物的形成过程,得到以下结论:
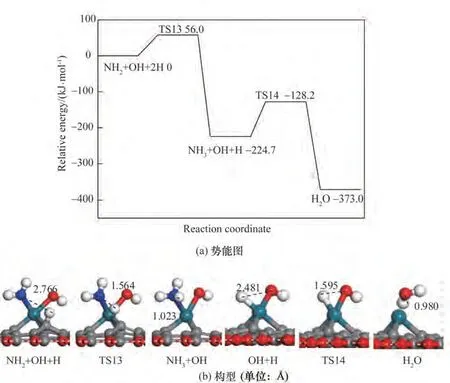
图5 NH3生成的势能图及对应的反应物、过渡态、产物的构型Fig.5 Potential energy diagram of correlated reactions of the formation of NH3 together with corresponding configurations of initial states,transition states and final states
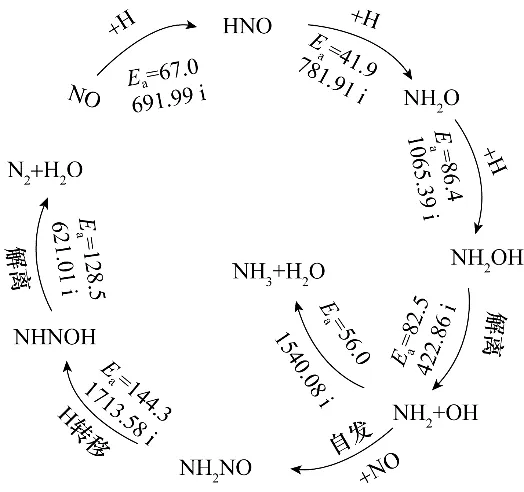
图6 N2与NH3生成的最优路径图及每一步基元反应的能垒(kJ·mol-1)和对应过渡态的虚频(cm-1)Fig.6 The formation pathways of N2 and NH3and the energy barrier(kJ·mol-1)for each elementary step and virtual frequency corresponding to the transition state(cm-1)
(1)在H2还原NO 的过程中,物种的稳定吸附位点主要在Pd 原子、Pd 与C 相连的桥位(B)以及与Pd相邻的C位,且反应也发生在这些位点。
(2)在Pd/SVG 上对NO 加氢形成HNO、NOH 和直接解离形成N 和O 的过程进行比较发现,NO活化的主要方式为NO 加氢形成HNO,仅需要克服67.0 kJ·mol-1的能垒,可见Pd/SVG对NO的活化能力强。
(3)阐明了Pd/SVG 上H2还原NO 的反应机理,生成N2的最优路径为NO 活化生成HNO,HNO 通过加氢、解离、结合、异构化过程经中间体NH2O、NH2OH、NH2、NH2NO 和NHNOH,最后经NHNOH 解离生成N2与H2O。NH3生成和N2的生成经过同样的中间体HNO、NH2O、NH2OH 和NH2,最后NH2加氢形成NH3。
(4)对生成N2和NH3的决速步骤能垒进行比较,生成N2的决速步骤为NH2NO 分子内氢转移生成NHNOH,所需要的活化能为144.3 kJ·mol-1;生成NH3的决速步骤为NH2O 加氢生成NH2OH,能垒为86.4 kJ·mol-1。可见Pd/SVG 上H2还原NO 过程中产物NH3的形成更容易。
猜你喜欢
北京航空航天大学学报(2022年5期)2022-06-06
兵工学报(2022年2期)2022-05-22
华东理工大学学报(自然科学版)(2022年2期)2022-04-29
浙江大学学报(理学版)(2021年6期)2021-12-02
中学课程辅导·教学研究(2021年8期)2021-07-14
化学工业与工程(2021年3期)2021-06-25
兵工学报(2021年4期)2021-06-19
兵工学报(2020年12期)2020-02-06
科学导报(2018年30期)2018-05-14
电脑知识与技术(2018年3期)2018-03-21
