
基于生物信息学方法的儿童急性不明确谱系白血病的潜在治疗靶基因筛选
2021-04-13 05:55吴任燕郭晓琳洪登礼
上海交通大学学报(医学版) 2021年3期
吴任燕,郭晓琳,洪登礼,陈 磊
1.上海交通大学基础医学院病理生理学系,细胞分化与凋亡教育部重点实验室,上海200025;2.上海交通大学医学院上海市免疫学研究所,上海200025
急性不明确谱系白血病(acute leukemia of ambiguous lineage,ALAL)是一种罕见的白血病类型,不能清楚地分为淋巴系和髓系,其发病率不到5%。根据2016 年世界卫生组织(World Health Organization,WHO)分类标准[1],ALAL 可分为多个亚型,包括急性未分化白血病(acute undifferentiated leukemia,AUL)、混合表型急性白血病(mixed phenotype acute leukemia,MPAL)等。尽管大多数白血病已经得到了很好的研究和治疗,但由于ALAL 的罕见性,目前普遍采用的是与急性髓系白血病(acute myeloid leukemia,AML)和急性淋巴细胞白血病(acute lymphoblastic leukemia,ALL)相同的治疗手段,无法对ALAL 进行针对性治疗,使得ALAL 相 较 于AML/ALL 治 疗 效 果 较 差[2-3]。因 此 探 究ALAL 发生和生存相关的基因和信号通路,有助于对ALAL发病机制的针对性研究,可为ALAL的临床治疗提供参考。
目前已有的研究主要针对ALAL的存活率、免疫分型以及相关microRNA 而开展。白血病生存分析[4-5]显示,大多数ALAL患者首选ALL的治疗方案;免疫表型分析[6]显示,早期T细胞前体淋巴细胞白血病和T/M-MPAL在免疫表型上相似,NOTCH1可作为T 细胞混合表型白血病的潜在治疗靶点;通过高通量测序,ALAL可以根据具有谱系特性的microRNA 表达特性被归类为AML 或ALL[7]。这些研究大多探讨了ALAL与其他白血病的谱系关系,但有关ALAL独特的基因表达谱、生存基因以及相关通路尚不清楚。高通量测序技术自问世以来,极大地推动了对于疾病标志物的研究进展。通过对公共数据库数据的生物信息学分析,我们可以对疾病的发病机制及治疗靶标进行预测,为疾病的机制研究和药物研发提供思路。
本研究利用小儿ALAL患者和健康对照组的表达谱数据(RNA-seq),对ALAL 中的差异表达基因(differential expressed genes,DEGs)进行筛选,利用基因本体数据库(Gene Ontology,GO) 和京都基因与基因组百科全书(Kyoto Encyclopedia of Genes and Genomes,KEGG)富集分析,探讨ALAL发病的生物学过程和相关信号通路;结合生存分析与蛋白互作网络,我们发现了在ALAL中独特的生存基因表达谱,鉴定了与ALAL生存相关的枢纽基因。本研究可为ALAL的临床诊断和治疗提供新的思路。
1 材料与方法
1.1 RNA-seq数据获得
基因表达数据来自于儿童ALAL 患者和儿童健康对照者的血液或骨髓的RNA-seq 数据。患者的基因表达数据是从UCSC xena 数据门户GDC 下载TARGET-ALL-P3数据集,该数据集由有效治疗应用研究(Therapeutically Applicable Research to Generate Effective Treatments,TARGET,https://ocg.cancer.gov/programs/TARGET) 计划生成。健康对照者的基因表达数据(GSE111459)从基因表达汇编数据库(Gene Expression Omnibus,GEO)下载。对异常样本进行过滤后,得到21 个健康儿童样本和69 个初诊儿童ALAL 样本分别作为健康对照组和ALAL组。纳入诊断样本和复发样本的数据用于不同类型白血病基因表达量比较,以TARGET-ALL-P2 的532 个ALL 样本为ALL 组,以TARGET-AML 的187 个AML 样本为AML 组,以TARGET-ALL-P3中的136个ALAL 患者样本为ALAL 完全组。所有患者的数据均可从癌症基因组图谱(The Cancer Genome Atlas,TCGA)获得。
1.2 前期处理及DEGs筛选
所有样本的表达量数据均被标准化为每百万片段计数(count per million,CPM)值以进行相关性分析和去除低测序质量样本。低表达基因(在所有样本中均CPM<1.5)和离群基因(<Q1-1.5×IQR或>Q3+1.5×IQR)被移除。利用Pearson相关分析方法对样本表达谱相关性进行分析。R语言limma软件包[8]用于鉴别ALAL标本与健康对照组之间的差异表达基因。采用Benjamini和Hochberg 修正法对多次试验进行修正,得到修正后的P 值(adjusted Pvalue)。为消除假阳性结果,DEGs的筛选标准为|log2Fold Change|(|log2FC|)>2且修正后的P<0.001。
1.3 DEGs的功能分析
为验证数据结果的可靠性,利用R 语言DOSE 软件包[9]进 行DEGs 的 疾 病 本 体 论(Disease Ontology,DO)[10]分 析。为 研 究ALAL 中 的 相 关 通 路,使 用clusterProfiler[11]的默认参数对上调和下调的DEGs 分别进行GO和KEGG富集分析。显著性筛选标准为P<0.05。
1.4 生存相关基因鉴定及其与AML/ALL的表达比较
69 例ALAL 患者中有48 个病例有总生存时间记录,并选择用于生存相关基因的鉴定。根据每个基因在所有样本中的平均表达量,48个样本被分为基因的高表达组和低表达组。生存分析采用R 语言的survival和survminer软件包进行,Cox 回归模型用于鉴定生存相关基因;以P<0.05,在表达下调的基因中相关系数>0或在表达上调的差异基因中相关系数<0,被认为是合理的ALAL生存相关基因。生存分析是针对与正常样本的差异基因进行,所得到的生存相关基因可能与小儿ALAL的致病相关,但这些生存相关基因的具体作用仍有待进一步的机制研究。
1.5 蛋白质-蛋白质相互作用网络构建及枢纽基因筛选
将生存相关基因导入GeneMANIA[12]构建蛋白质-蛋白质相互作用网络(protein-protein interaction network,PPI),该网络中总共包含22 个ALAL 生存相关基因以及100 个与这些基因具有功能或表达相关性的间接相关基因。利 用Cytoscape[13]绘 制PPI 网 络 图 谱,并 利 用Cytohubba[14]计算网络图中每一个基因的最大群体中心度(maximal clique centrality,MCC),得分值最高的前10个基因,被认为是PPI网络中的枢纽基因。
1.6 统计学方法
2 结果
2.1 数据质控
由于测序技术的特性,我们需要对不同来源的测序数据进行质控,以提高最后分析结果的准确性。在去除低质量样本和离群基因后,在原始数据中筛选出总共90个样本的19 213 个基因的表达值进行后续分析,其中包括21 个健康对照组和69 个儿童ALAL 组的样本。标准化前后的表达值如图1A 所示。在ALAL 组和对照组中的基因表达量显示出较高的内部样本相关性(图1B),证明筛选得到的数据内部表达谱具有均质性,可以用于后续分析。样本编号与原始数据编号的对应关系见附表1。

图1 样本数据质控Fig 1 Quality control of sample data

表1 生存相关基因在不同类白血病患者及健康对照者中的表达值(log2CPM)Tab 1 The log2CPM value of survival-related genes in the control and different leukemia types
2.2 DEGs筛选与功能分析
为进一步探究儿童ALAL 的异常基因表达谱,利用limma 软件包对ALAL 组和健康对照组的基因表达谱进行差异分析;选择|log2FC|>2、校正后的P<0.001 为条件进行基因筛选,去除假阳性基因,共鉴定得到4 053 个基因,其中上调基因1 844 个,下调基因2 209 个。基于所有DEGs 的DO 分析表明造血系统疾病是最显著富集的疾病(图2A),与我们的数据特征相符,进一步证明我们筛选使用的样本数据和差异分析的合理性。对上调和下调的差异基因分别进行GO 和KEGG 富集分析,找寻与ALAL 发病相关的信号通路,各组显著富集的前10 个生物学过程和信号通路如图2B 及图2C 所示。ALAL 组中与免疫相关的生物学进程、造血细胞谱系和细胞黏附分子相关的信号通路明显下调,而细胞周期、有丝分裂以及剪接相关的生物学进程在ALAL 组中明显富集。
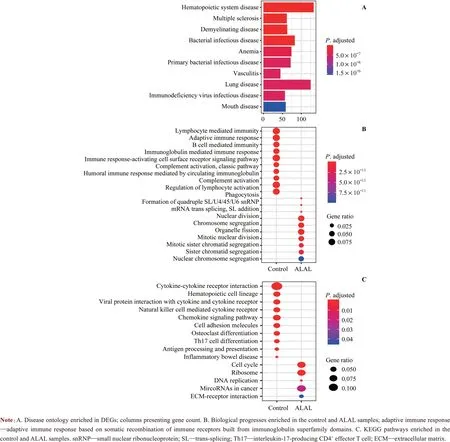
图2 差异基因的功能分析Fig 2 Functional analysis of DEGs
2.3 生存相关基因筛选及表达谱的比较
利用ALAL 患者的总体生存期资料,我们从4 053 个差异表达基因中鉴定出31 个ALAL 生存相关基因。其中13 个基因在ALAL 组中表达量增加,与生存呈负相关,被认为是负生存相关基因;18 个基因在ALAL 组中表达降低,与生存呈正相关,被认为是正生存相关基因。
此外,为了研究这些基因是特异在ALAL 中表达变化还是白血病的普遍差异基因,我们在ALL 组以及AML组的表达谱中对这些基因进行了比较分析。结果表明,与ALAL 生存相关的基因,在ALL 组以及AML 组中呈现出同样的差异表达趋势,相对于健康组,在ALAL 完全组中表达上调的基因同样在ALL 组和AML 组中表达上调,但这些基因的表达量在各类型白血病之间仍然存在显著差异(表1)。这些基因中也有一些“偏向性”表达特例,SMARCA5-AS1 在AML 组中的表达与对照组相比差异不具有统计学意义(P=0.151),但在ALAL完全组和ALL 组中表达较对照组有明显增加(均P=0.000)。ZNF135在ALAL完全组和AML组中的表达值均低于对照组,且差异具有统计学意义(均P=0.000),但在ALL 患者中表达量无明显变化(P=0.073)。这些数据为研究ALAL的独特生存特征及治疗方案的选择提供了线索。
2.4 PPI网络构建以及枢纽基因识别
枢纽基因是在PPI网络中具有紧密的相互联系,发挥核心作用的一群基因。为了进一步筛选出在ALAL 患者生存中发挥关键作用的枢纽基因,我们将所有生存相关基因导入到GeneMANIA 中,构建了PPI 网络(图3A)。GeneMANIA 中的GO 富集分析显示细胞趋化性和白细胞迁移相关的生物学过程在这个相互作用网络中显著富集,提示ALAL 患者的生存与细胞趋化性及白细胞迁移相关。在这个PPI 网络中MCC 得分最高的前10 个基因被认为是这个PPI 网络中的枢纽基因(图3B)。在这10 个基因中,只有CXCL8 和LMNA 与ALAL 的生存相关且在ALAL 组中表达量发生变化,被鉴定为ALAL 的生存相关枢纽基因。在ALAL 组和健康对照组中CXCL8 和LMNA 的生存曲线和表达谱如图3C 和图3D 所示,CXCL8 和LMNA 均为负生存相关基因,在ALAL组中表达上调,伴随ALAL患者总生存期下降。

图3 ALAL生存相关枢纽基因鉴定Fig 3 Survival-related hub genes identification and its character.
3 讨论
本研究通过生物信息学分析的方法,将基因表达数据与临床生存数据结合,对ALAL 的生存相关枢纽基因进行筛选。为使得研究结果更加准确,我们首先对公共数据库的数据进行了筛选,得到21 个健康样本(GSE111459) 和69 个ALAL 样 本(TARGET-ALL-P3)的共19 213 个基因表达数据。基因表达谱的差异分析显示在ALAL组中有1 844个基因表达上调,2 209个基因表达下调,揭示了ALAL 的异常基因表达谱。疾病本体分析的结果显示造血系统疾病在差异基因中高度富集,进一步验证了我们的数据以及差异分析的可靠性。GO 以及KEGG富集分析表明在ALAL组中上调表达的基因主要富集细胞周期以及有丝分裂的相关进程,而下调的差异基因在造血细胞谱系相关通路和免疫相关进程中富集。白血病的主要特征为造血干细胞的恶性增殖和分化障碍,因此细胞周期及有丝分裂进程的上调,造血细胞谱系相关进程的下调,符合白血病的基本特性。但目前尚未有研究指出ALAL 中免疫相关进程的下调,因此本研究中找到的通路可能为ALAL的免疫学治疗提供理论依据。
结合临床生存记录,我们从ALAL 的差异基因中筛选出了31 个与生存相关的基因。由于目前ALAL 缺乏明确的靶向治疗方案,ALAL 的治疗普遍是采用ALL 或者AML 的治疗方案进行,但这些治疗往往带来较差的预后效果[2-3],因此我们将ALAL 的生存相关基因表达谱与ALL/AML 进行比较,探究特异在ALAL 中异常表达的生存基因。结果显示大多数基因在ALAL、AML 和ALL 中具有共同的上下调趋势,这些ALAL 的生存相关基因,在AML/ALL 中同样差异表达,具有白血病致病的普遍性。但这些基因在各个白血病类型之间的表达谱存在明显差异,ALAL 的生存相关基因表达量大多数介于ALL和AML 之间,表现出与ALAL 生存相关的特殊基因表达谱。此外我们发现,在这些基因中存在一些明显的“偏向”调节的情况,如SMARCA5-AS1 在ALAL 和ALL 中表达增加,但在AML 中没有改变;ZNF135 在ALAL 和AML 中表达下调,但在ALL 中没有改变。这些数据可能为ALAL治疗方案的选择提供借鉴作用。
此外,通过PPI网络图谱的构建,我们进一步筛选出CXCL8 和LMNA 作为ALAL 生存相关的枢纽基因。CXCL8 又称IL-8,是CXC 趋化因子家族的成员。CXCL8可从白细胞和非白细胞的体细胞中释放,在炎症中经常上调表达,并在白细胞诱导迁移和释放反应中发挥作用[15]。近期的研究表明,白血病细胞广泛释放CXCL 和CCL 趋化因子,尤其是高水平地释放CXCL8[16];CXCL8与AML 的复发有关,CXCL8 的敲除导致G0/G1 细胞周期阻滞、凋亡和细胞外调节蛋白激酶1/2 信号通路失活[17];CXCL8 可与CXCR1/2 相互作用,调节细胞增殖和分化,介导肿瘤的发生和发展[18-19]。然而,CXCL8 在ALAL 中是否发挥作用尚不清楚。在本研究中,CXCL8 在ALAL中表达上调;并在ALAL 患者的生存中充当枢纽基因,ALAL 患者中富集与细胞周期相关的信号通路,CXCR1/2在ALAL 中表达下调,结合已有报道,我们推测CXCL8可能通过参与细胞周期的进程介导了ALAL 的发生和发展,而并非通过与CXCR1/2 相互作用参与ALAL 的发生。LMNA 编码核包膜蛋白lamin A 和lamin C,主要参与染色质组织、核组装和端粒动力学,与涉及凋亡和存活的Caspase 级联途径有关。LMNA 突变引起多种疾病[20-21],但在白血病中对LMNA 的研究非常少。此前,有报道称LMNA 在活性T 细胞中被诱导表达[22],但LMNA 在造血细胞中的表达情况尚不明确。通过高通量测序数据,我们在对照组和ALAL 组中均检测到LMNA,并且LMNA 在ALAL患者中的表达明显高于对照组。我们的结果首次提出LMNA在ALAL的发生和生存中发挥作用。
综上所述,我们对儿童ALAL 病例的独特基因表达谱进行了研究,确定了参与ALAL 发生的信号通路及生物学进程,并将ALAL 生存相关基因的表达谱与ALL 和AML 的表达谱进行了比较,为研究ALAL 的独特生存特征和现有治疗方案的选择提供了线索。基于生存期分析找到了ALAL生存相关枢纽基因,可能作为潜在的ALAL治疗靶点,但这些枢纽基因的功能仍有待进一步验证。
猜你喜欢
人人健康(2022年2期)2022-11-21
房地产导刊(2020年8期)2020-09-11
父母必读(2019年4期)2019-09-09
家庭影院技术(2019年4期)2019-04-17
当代陕西(2018年12期)2018-08-04
当代陕西(2018年12期)2018-08-04
中国中药杂志(2017年15期)2017-08-30
中国中药杂志(2017年15期)2017-08-30
江苏农业科学(2017年5期)2017-04-15
湖北农业科学(2014年3期)2014-07-21
