
分散型催化剂作用下噻吩类化合物的临氢转化规律
2021-05-20 08:25侯焕娣申海平代振宇
石油学报(石油加工) 2021年2期
侯焕娣, 赵 毅, 董 明, 申海平, 代振宇, 龙 军
(中国石化 石油化工科学研究院,北京 100083)
近年来,环保法规日益严格,对石油产品的硫含量要求越来越低,欧Ⅴ汽、柴油产品标准中要求硫质量分数小于10 μg/g[1]。柴油深度加氢脱硫主要是通过加氢使油品中的含硫化合物发生氢解而转化成相应的烃和H2S,从而使其中的硫原子得以脱除。原油中的硫80%集中在渣油中,因而渣油加工中脱硫是一个主要目的。崔文龙等[2]以轮古常压渣油为研究对象,研究了热转化过程中类型硫的转化规律,结果表明,在热反应过程中噻吩硫同时存在裂解和缩聚反应,但需在较高反应温度下进行。任亮等[3]以海南炼化渣油为原料,采用第三代渣油加氢处理RHT催化剂进行深度脱硫的RHT工艺研究,获得实现装置长周期稳定生产硫质量分数不大于0.2%的加氢渣油优化工艺条件和催化剂级配方案。Hao等[4]以介孔分子筛为载体,制备了不同Mo-Co含量的加氢脱硫(HDS)催化剂并进行催化脱除二苯并噻吩性能研究,考察催化剂孔体积、比表面积以及酸性对脱硫率的影响,结果表明,介孔分子筛大比表面积使Co、Mo活性组分高度分散在载体表面,载体与金属之间较适中的相互作用有利于活性组分的还原和硫化,含有沸石结构单元使其具有更多的酸中心,有利于提高加氢脱硫反应活性。渣油浆态床加氢技术是采用高分散度的分散型催化剂实现渣油高效改质的新技术,其原料适应性广、渣油转化率高,被视为实现渣油轻质化的有效途径。现有的众多研究[5-9]集中在催化剂及工艺条件对反应的影响方面,对于分散型催化剂作用下含硫化合物的脱硫途径及转化规律研究较少。笔者制备出高分散度纳米尺寸的分散型钼基催化剂RDC-Mo,选取不同结构的噻吩类含硫化合物(噻吩、苯并噻吩和二苯并噻吩)作为反应物,同时采用实验和分子模拟计算2种方法,研究了分散型催化剂作用下不同结构含硫化合物的临氢转化反应规律。
1 实验部分
1.1 原料和试剂
实验用的模型化合物噻吩、苯并噻吩及二苯并噻吩均为北京化学试剂公司产品,通过GC-MS分析其质量分数均大于98%;溶剂四氢萘、二甲基二硫醚(DMDS),均为分析纯,北京试剂公司产品;柴油取自中国石化北京燕山分公司;液体有机钼,实验室自制合成。
1.2 纳米分散型催化剂的制备
以液体有机钼作为前驱体,柴油为硫化油,二甲基二硫醚为硫化剂,在反应温度350 ℃下反应30 min,得到的产物经过过滤、抽提、烘干,制备出高度分散纳米尺寸分散型催化剂RDC-Mo。
1.3 纳米分散型催化剂的表征
采用日本理学TTR3 X-射线衍射仪进行催化剂物相晶体结构的表征,光源为CuKα辐射,管电压40 kV,管电流250 mA,扫描范围5°~70°,扫描速率为4 °/min。采用Tecnai G2F20S-TWIN高分辨透射电镜进行TEM表征,电子枪为LaB6,加速电压200kV。采用ESCALab250型X射线光电子能谱仪(Thermo Scientific)表征催化剂样品表面组成和金属价态,激发源为单色化AlKαX射线,能量为1486.6 eV,功率为150 W,分析时的基础真空为6.5×10-8Pa。采用XPSPERK Version 4.0对Mo3d能谱进行拟合分峰,根据峰面积计算得到相应价态Mo含量。
1.4 分散型催化剂催化噻吩类化合物临氢热转化实验
采用100 mL微型反应釜进行热转化和分散型催化剂作用下的噻吩类模型化合物转化实验,其中热转化、催化临氢热转化分别在N2、H2气氛下,在初始压力9 MPa、反应温度420 ℃、反应时间 60 min 条件下进行。具体实验步骤如下:在微型反应釜中分别添加1 g噻吩类化合物(噻吩、苯并噻吩及二苯并噻吩)、9 g溶剂四氢萘、0.3%~1%(质量分数)分散型催化剂RDC-Mo(催化临氢实验),然后将反应釜密封,向反应釜中充入9 MPa的H2或N2进行气密性测试,气密性合格后启动工作站,以升温速率10 ℃/min升温至反应温度,反应3 h后结束,关闭加热。待反应釜内温度降至室温,收集气体并通过Ritter气表计量气体体积,采用气相色谱(GC)进行组成分析,根据气体组成分析结果获得气体平均相对分子质量,计算得到气体质量和气体收率;收集液体产物并称量计算液体产物收率,同时采用美国Agilent Technologies公司生产的7890A-5975C气相色谱-质谱(GC-MS)联用仪进行液体产物组成分析。色-质联用条件为:色谱柱规格30 m×250 μm×0.25 μm,HP-5MS,进样口温度300 ℃,进样量0.1 μL,分流比400/1;程序升温条件为:在5 min内升温至50 ℃,然后以升温速率6 ℃/min升温至300 ℃;载气流速1 mL/min;离子源温度220 ℃,质量扫描范围50~700,EI电离源;FID:加热器温度350 ℃,H2流量30 mL/min,空气流量300 mL/min,氦气流量25 mL/min。
实验数据处理中假设溶剂不参与反应,根据GC分析结果、结合MS数据确认每一种产物的结构和含量,扣除溶剂,计算原料(噻吩、苯并噻吩及二苯并噻吩)的转化率、直接脱硫产物收率以及噻吩类化合物芳环加氢氢化噻吩化合物收率,其计算公式如式(1)~(3)所示。
x=100%-wF×yL×100%
(1)
yDS=(yG×wH2S+yL×wHC)×100%
(2)
yHT=yL×wHT×100%
(3)
式中:x为原料转化率,%;yL为液体产物收率,%;yG为气体产物收率,%;yDS为直接脱硫产物收率,%;yHT为氢化噻吩类化合物收率,%;wF为液体产物GC-MS分析结果中扣除溶剂后原料的质量分数,%;wH2S为气体组成中H2S质量分数,%;wHC为液体产物GC-MS分析烃类化合物质量分数之和,%;wHT为液体产物中氢化噻吩类化合物质量分数之和,%。
1.5 分子模拟软件及方法
采用Accelrys公司Materials Studio6.0软件中的DMol3模块进行噻吩类化合物临氢反应脱硫过程反应化学分子模拟。对反应过程的研究,主要是通过对反应的过渡态进行搜索,求取反应能垒和反应热。计算的具体过程为:首先利用Materials Studio6.0构建反应物及产物的结构模型,利用基于密度泛函理论的模块DMol3进行结构优化,反应物及产物的结构优化计算成功之后,采用LST/QST方法来搜索过渡态,过渡态搜索计算完成后,通过振动频率分析确认过渡态是否正确;最后计算反应能垒。
2 结果与讨论
2.1 分散型催化剂结构组成的表征结果
分散型催化剂RDC-Mo与标准MoS2晶体的X射线衍射图如图1所示。由图1可知:分散型催化剂RDC-Mo在2θ为14°、32.6°、39.5°、60.4°处出现MoS2的特征峰,但与标准MoS2晶体的X射线衍射谱图相比,RDC-Mo的X射线衍射谱图中特征峰宽而缓,根据Kim等[10]研究结果,这是因RDC-Mo催化剂粒度较小所致。运用谢乐公式计算RDC-Mo晶粒尺寸为3.63 nm。
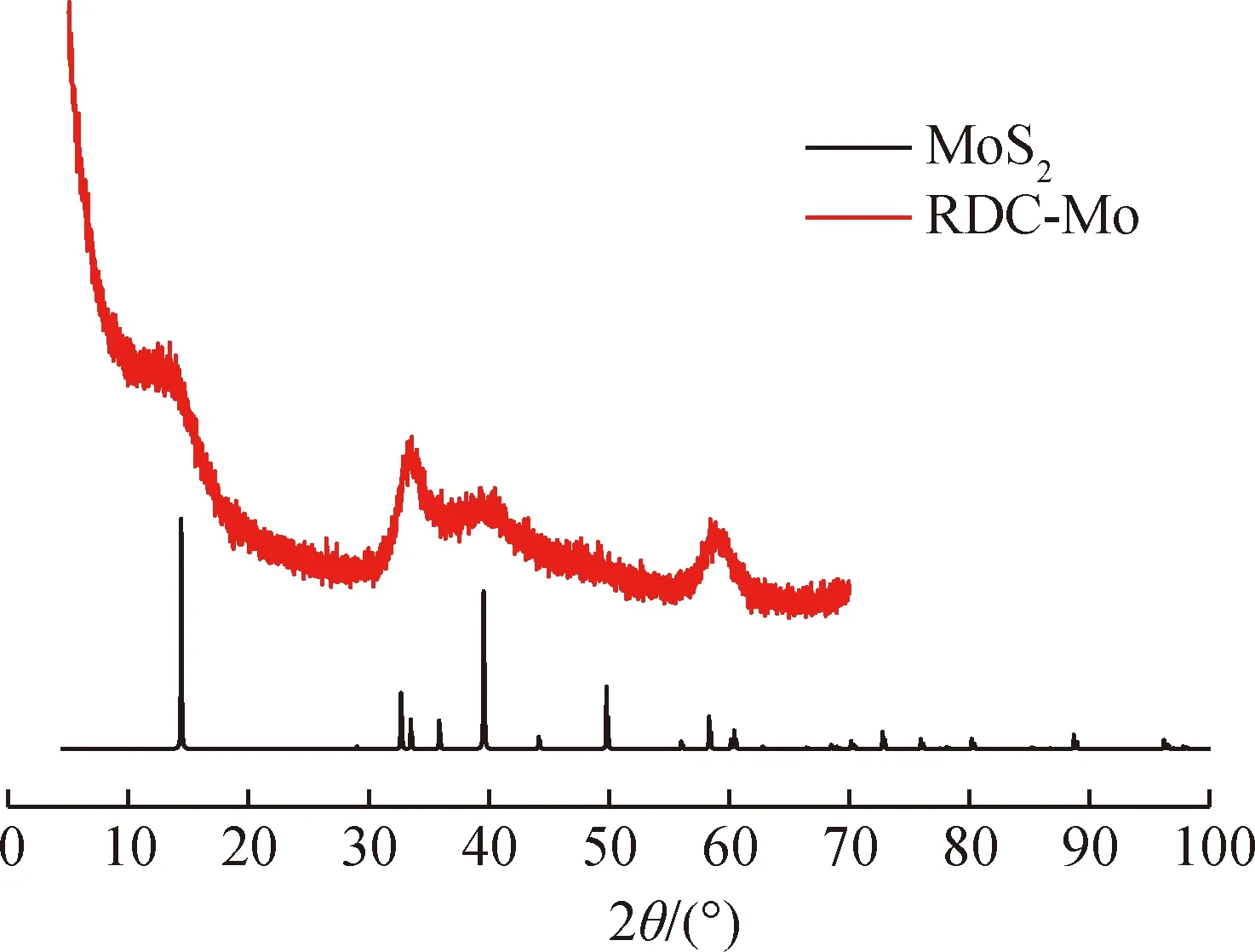
图1 分散型催化剂RDC-Mo和标准MoS2的X射线衍射谱图Fig.1 X-ray diffraction analysis of dispersed catalystRDC-Mo and standard MoS2
图2为分散型催化剂RDC-Mo的外观形貌及活性相HRTEM照片。由图2可知:分散型催化剂RDC-Mo呈粉末状;其活性相分散均匀,以单层和双层分散为主,活性相的尺寸为纳米级,这与其XRD表征结果吻合。相比于常规微米或毫米级别负载型催化剂,分散型RDC-Mo催化剂由于尺寸更小,且无载体,不涉及孔道扩散,因此其与H2、噻吩类化合物的可接近性更强。

图2 分散型催化剂RDC-Mo的外观形貌及HRTEM照片Fig.2 Morphology and HRTEM images of the dispersed catalyst RDC-Mo(a) Appearance of RDC-Mo; (b) TEM of RDC-Mo
表1为分散型催化剂RDC-Mo的X射线光电子能谱分析的元素组成结果。从表1可知:RDC-Mo催化剂主要由C、Mo、S、O元素组成;通过对Mo 3d 能谱进行拟合分峰,根据峰面积计算得到+4价态Mo占催化剂中总Mo的质量分数为94.08%,表明RDC-Mo催化剂硫化度很高;根据S2-/Mo4+摩尔比计算得到RDC-Mo催化剂n(S)/n(Mo)=1.76,表明RDC-Mo催化剂并非是MoS2化学计量的,催化剂中含有更多的空缺位(即活性位)。

表1 RDC-Mo催化剂的主要元素组成Table 1 Main element composition of RDC-Mo catalyst
2.2 不同结构噻吩类化合物热转化和催化临氢反应结果对比
以四氢萘做溶剂,质量分数均为10%的噻吩、苯并噻吩及二苯并噻吩分别作为反应物,在反应温度420 ℃、初始压力9 MPa、反应时间60 min条件下进行噻吩、苯并噻吩及二苯并噻吩热转化反应(N2气氛、无催化剂)以及添加质量分数为1%的分散型催化剂RDC-Mo临氢条件下的催化临氢热转化反应,不同反应条件下噻吩类化合物转化率列于表2。
由表2可知:噻吩、苯并噻吩及二苯并噻吩3种噻吩类化合物,N2气氛下无催化剂的热转化反应转化率均为0;添加1%分散型催化剂RDC-Mo的催化临氢反应体系中,噻吩类化合物转化率显著提高,3种不同结构含硫化合物转化率由大到小顺序为苯并噻吩、噻吩、二苯并噻吩。

表2 不同结构噻吩类化合物热转化反应及催化临氢热转化反应的转化率(x)Table 2 Conversions ratios (x) of thermal and catalytic hydro-thermal reaction of different structural thiophenes compounds
在分散型催化剂RDC-Mo作用下,在反应温度420 ℃、H2初压9 MPa、反应时间60 min条件下,不同结构噻吩类化合物催化临氢热转化反应主要产物如表3所示。在实验条件下,噻吩类化合物发生两类反应,即加氢脱硫生成H2S和烃类化合物、加氢饱和生成氢化噻吩类化合物。由表3可知,3种不同环数噻吩类化合物的加氢脱硫产物收率都远高于加氢饱和产物氢化噻吩类化合物,表明在分散型催化剂RDC-Mo作用下,相比于加氢饱和生成氢化噻吩,噻吩类化合物更容易发生直接脱硫生成H2S和烃类化合物。同时还可以看到,不同环数、不同结构噻吩类化合物加氢饱和产物收率不同,其中二苯并噻吩的加氢饱和产物收率最高。

表3 分散型催化剂RDC-Mo催化噻吩类化合物的临氢热转化结果Table 3 Experimental results of thiophenes hydro-thermal cracking with dispersed catalyst RDC-Mo
2.3 反应条件对二苯并噻吩催化临氢热转化反应的影响
由表2可见,在催化临氢热转化反应中3种噻吩类化合物中的二苯并噻吩的转化率较低,仅有62.70%。为了研究二苯并噻吩适宜的临氢转化条件,在H2初压9 MPa、分散型催化剂RDC-Mo质量分数0.3%条件下,分别考察了不同反应温度、不同反应时间二苯并噻吩的临氢转化结果,示于图3 和图4。

xDBT—DBT conversion图3 不同反应温度(T)下二苯并噻吩(DBT)化合物催化临氢热转化反应结果Fig.3 Experimental results of dibenzothiophene (DBT) catalytic hydro-thermal reaction at different reaction temperaturesReaction conditions: t=180 min; p=9 MPa; w(RDC-Mo)=0.3%

xDBT—DBT conversion图4 不同反应时间(t)下二苯并噻吩(DBT)化合物催化临氢热转化反应结果Fig.4 Experimental results of dibenzothiophene (DBT) catalytic hydro-thermal reaction under different reaction time (t)Reaction conditions: T=430 ℃; p=9 MPa; w(RDC-Mo)=0.3%
由图3、图4可知,随着反应温度升高、反应时间延长,二苯并噻吩转化率均会提高,当反应温度升至430 ℃、反应时间延长至180 min时,二苯并噻吩的转化率可达98%。这主要是因为反应温度升高,反应速率加快,同时温度升高,给予体系的能量增多,均使得二苯并噻吩转化率提高。
3 噻吩类化合物催化临氢热转化脱硫计算化学结果
为了探寻分散型催化剂RDC-Mo作用下,含硫化合物催化临氢条件下转化率大幅提高的原因,以及不同结构噻吩类含硫化合物催化临氢热转化反应性能的差异,以二苯并噻吩为含硫化合物模型分子,采用密度法函数的DMol3模块进行了催化临氢热转化的反应化学计算。
3.1 二苯并噻吩催化临氢热转化脱硫反应机理
计算了二苯并噻吩分子C—S键键能和键长,如图5所示。由图5可见,二苯并噻吩分子C—S键能最低为342.5 kJ/mol,C—S键键长0.176 nm。

Bond energy unit: kJ/mol图5 二苯并噻吩分子结构、键长和键能Fig.5 Dibenzothiophene molecular structure,bond length and bond energy
在催化临氢热反应体系中,H2经催化剂活化形成氢自由基,会攻击二苯并噻吩苯环6个不同位置的碳,使其发生加氢饱和反应。经过计算获知,氢自由基进攻二苯并噻吩芳环上碳反应能垒很低,仅有30 kJ/mol,如图6所示。
当氢自由基攻击二苯并噻吩苯环6个不同位置的碳时,会改变芳环碳的杂化方式,从而会使C—S键键长发生变化。图7为氢自由基进攻二苯并噻吩芳环不同位置碳C—S键长的变化。从图7可知,只有当·H加在C 4位上时,C—S键键长才明显增长(0.1760 nm→0.1891 nm),进攻其他位置碳,对C—S键的键长影响不大,甚至使其缩短。

图6 氢自由基进攻二苯并噻吩芳环碳反应能垒Fig.6 Energy barrier of hydrogen radicals attacking dibenzothiophene aromatic ring carbon

1-6 indicate the carbons at different positions of the dibenzothiophene aromatic ring; Bond length unit: nm图7 氢自由基进攻二苯并噻吩不同位置碳C—S键长的变化Fig.7 Change of C—S bond length when hydrogen radical attacking different carbons in dibenzothiophene
在C 4位加氢后,5位形成自由基,此时C—S键被削弱;同时C 4位的σ成键方式由C sp2-S sp2变为C sp3-S sp2;由于C 4位杂化方式的改变,S的pz轨道不能再与芳环的大π键发生共轭。基于上述3个原因,C—S键的键长明显增长,键能大幅降低,降为12.7 kJ/mol(见图8),此时C—S键常温下很容易发生断裂。

图8 氢自由基进攻C4二苯并噻吩C—S键断裂反应Fig.8 Dibenzothiophene C—S bond rupture when hydrogen free radicals attacking C4 position
C 4位加氢后,如果H·进攻S,也会使得C—S键断裂,此时断裂能垒很低,仅有11.8 kJ/mol(见图9);同时看到其逆反应能垒很高,表明C 4位加氢后,氢自由基再进攻S,C—S键很容易发生断裂,且不易发生逆反应。
图8、图9所示的反应路径表明,含硫杂环烃芳环定向加氢-C—S键弱化-高选择性断裂的反应路径可促使C—S键在低温下发生断裂,该路径正是催化临氢反应体系,采用高度分散的纳米催化剂RDC-Mo,使得催化剂、氢气与二苯并噻吩特殊碳,即与S相连的碳(如C 4位)具有较高的可接近性,这是使噻吩、苯并噻吩、二苯并噻吩具有高裂化转化率的原因。

图9 C4加氢饱和氢自由基进攻S二苯并噻吩C—S键断裂反应Fig.9 Dibenzothiophene C—S bond rupture when hydrogen radicals attacking C4 position and sulfur hydrogenation taking place
3.2 不同结构噻吩类化合物催化临氢热反应的差异分析
噻吩、苯并噻吩及二苯并噻吩的结构式及特殊碳如图10所示。由二苯并噻吩反应化学研究可知,分散型催化剂RDC-Mo催化含硫杂环烃芳环定向加氢-C—S键弱化-高选择性断裂的反应路径是使其临氢转化脱硫率显著提高的原因。由图10可知,噻吩、苯并噻吩及二苯并噻吩的特殊碳都是与S相邻的碳(图10中标*碳),而活性氢进攻该位置碳的概率分别为2/4(即50%)、2/8(即25%)和2/12(即16.7%),因而从活性氢进攻特殊碳的概率数据可知,发生定向加氢-C—S键选择性断裂反应几率由大到小顺序为:噻吩、苯并噻吩、二苯并噻吩。

图10 噻吩、苯并噻吩及二苯并噻吩的结构式及特殊碳Fig.10 Carbon positions connected with sulfur in thiophene, benzothiophene and dibenzothiophene and special carbon
模拟噻吩、苯并噻吩在硫化钼活性中心的吸附能,噻吩的吸附能为-55 kJ/mol,苯并噻吩吸附能为-70 kJ/mol(负值表示吸附是放热反应),相比于噻吩,苯并噻吩吸附能更大,在反应过程中更容易吸附到活性中心上发生反应。计算噻吩和苯并噻吩特殊碳定向加氢后C—S键长,结果见图11。由图11 可知,相比于噻吩,苯并噻吩特殊碳被定向加氢后,其C—S键增长更多,更容易断裂,这与其反应结果(见表2)规律一致。
4 结 论
(1)相比于热转化,分散型催化剂RDC-Mo催化临氢热反应可显著提高噻吩、苯并噻吩及二苯并噻吩的转化率。

Bond length unit: nm图11 噻吩、苯并噻吩特殊碳定向加氢后的C—S键长Fig.11 C—S bond length after hydrogen radical attacking the carbon connecting with sulfur in thiophene and benzothiophene
(2)采用纳米分散型催化剂RDC-Mo催化二苯并噻吩临氢热转化,其活化H2形成的氢自由基能够有效接触并进攻噻吩类化合物特殊碳(即与S相连的碳),改变其杂化形式,促使C—S键断裂能垒大幅下降,使其发生低温断裂;这正是分散型催化剂作用下的催化临氢体系显著提高含硫化合物裂化转化率的原因。
(3)受特殊碳定向加氢的概率、在活性中心MoS2吸附难易以及特殊碳定向加氢后C—S键键长变化3方面影响,不同结构噻吩类化合物转化率由大到小的顺序为苯并噻吩、噻吩、二苯并噻吩。
猜你喜欢
分子催化(2022年1期)2022-11-02
武汉工程大学学报(2022年4期)2022-08-26
中草药(2022年9期)2022-05-06
四川化工(2022年1期)2022-03-11
石油炼制与化工(2021年12期)2021-12-14
天然气与石油(2021年5期)2021-12-05
石油炼制与化工(2021年7期)2021-07-14
烟台大学学报(自然科学与工程版)(2021年1期)2021-03-19
科学与财富(2020年18期)2020-09-09
天然产物研究与开发(2018年2期)2018-04-04
