
药物结晶中的经典与非经典结晶路径
2021-05-20 10:08宋舒虹姚昌林曲亚倩陶绪堂
人工晶体学报 2021年4期
宋舒虹,姚昌林,王 蕾,曲亚倩,陶绪堂
(山东大学,晶体材料研究所,晶体材料国家重点实验室,济南 250100)
0 引 言
结晶在地球化学、生物及合成材料中占据核心地位,长期以来一直受到研究人员的广泛关注,如在化学、制药和食品工业,结晶实现了对产品的造粒与提纯。尤其在制药领域,超过70%的药物都是以固体形式存在的[1],包含了多晶型、水合物、盐和溶剂化物,除此之外,目前一些药物的非晶态形式也得到了广泛应用。结晶作为一个极其重要的步骤,深刻影响着药物的上、下游加工工艺,其中药物晶型是影响固体药物质量和疗效的重要因素。药物晶型不同使得药物的溶解度、溶出速率、稳定性等理化指标有很大差异,直接影响到药物的生物利用度、安全性和疗效。因此药物晶型研究是目前科学界及制药工业界的重要研究内容和世界性前沿课题[2]。然而,目前晶型调控存在各种问题,如晶型有时会突然“消失”,即晶型调控重复性、可控性差的问题,晶型失控现象给企业带来严重后果,使产品研发迟滞、商业生产中断。1998年,美国雅培公司的HIV蛋白酯酶抑制剂上市两年后,发现相同的制剂工艺下,原料药晶型“消失”,转晶成了溶解度更小的更稳定晶型,最终导致已经上市的药品撤出市场,损失达上亿美元[3]。“伴随结晶”也是晶型调控中常见的现象,它会影响晶型的纯度,增加晶型调控的难度[4]。究其原因,是对晶型的成核和生长过程研究不足,对晶体成核和生长途径及机理不清楚,不能准确实施控制。因此加强药物晶体的成核和生长路径研究,了解结晶过程的前驱体相,对于调控药物分子晶型,实现晶型的主动调控有重要理论指导意义[5]。
经典成核理论[6-8](classical nucleation theory, CNT)认为:结晶过程中存在一个临界晶核尺寸,超过该尺寸,晶核通过单体的依次添加实现生长,最终形成结构有序的晶体。然而,越来越多的实验现象表明,在一些物质的结晶过程中,单体首先转变为低聚物、复合物、富溶质液滴、纳米晶粒等中间态[9-11],以该前驱体作为生长单元实现晶体的生长,研究人员将该过程归结为非经典的结晶路径(non-classical crystallization, NCC)。在药物结晶领域,不同晶型药物结晶的中间过程是怎样的?是否存在中间体?中间体如何继续长大并进一步发展成为晶体?以上均是目前药物结晶机理研究的重要内容,也是难点所在。
1 溶液中的经典结晶路径
CNT认为,结晶是一个由分子团簇到晶核再到晶体的变化过程,而晶核是结晶前期溶液中的分子通过自组装形成的一种有序聚集体,药物的晶型是由成核过程所决定的。成核与化学反应不同,有序聚集态的存在与否取决于它们的尺寸,晶核产生时需要消耗一定的自由能来形成新的界面,可利用的自由能取决于过饱和度。因此在过饱和溶液中,将分布一些稳定的聚集态,根据所采用的过饱和度将存在一个临界尺寸,超过这个尺寸,聚集态继续长大,低于该临界尺寸则处于不稳定状态。过饱和度越高,临界尺寸越小,越容易成核,产生的晶体数量也越多[12]。
1.1 基于CNT的成核热力学和动力学
1897年,奥斯特瓦尔德提出了著名的奥斯特瓦尔德阶段定律(Ostwald’s law of stages)[13],指出结晶过程中首先出现的是最不稳定晶型,随后出现稳定性依次提高的其他晶型,不同晶型出现的先后顺序与它们自身的稳定性有关。奥斯特瓦尔德阶段定律是在药物结晶实验中常用的指导原则, 是经过一系列实验数据总结得到的一个基本规律,是对晶型成核难易程度的基本判断依据。然而这个规则并不一直有效。例如CaCO3各个晶型在模拟体液中的转化并不符合奥斯特瓦尔德阶段定律[14]。对乙酰氨基酚从水中结晶得到的是稳定晶型I[15]。奥斯特瓦尔德时期,由于饱和溶液或者熔体中存在亚稳区间或过冷现象,即热滞后现象,人们由此推断成核存在势垒。直到Volmer和Gibbs等阐明了决定成核过程的参数,进一步发展了成核动力学过程理论,成核势垒才真正被人们理解[16-17],即CNT。
CNT是目前唯一可用于解释晶体成核过程的比较成熟的理论,并在后续发展中不断得到完善[18],该理论指出,晶体的成核速率J可简单地表达为:
(1)
式中:J为晶体成核速率,即单位时间内单位体积过饱和溶液中产生晶体的数量;S为过饱和度,是评价过饱和体系脱离平衡态程度的指标;A、B为常数,根据B/ln2S=W/kT又定义了成核功,即成核势垒。
由成核速率表达式可知,由于存在成核势垒,因此溶液结晶中通常存在一个亚稳浓度区间,在该区间内,尽管溶液过饱和,自发成核可以近似忽略,超过该区间,过饱和体系是不稳定的。这也从热力学上解释了实际实验中观察到成核延迟发生的动力学现象。另外,Mullin等[19]还提出了“生长死区(growth dead zone)”的概念,通常出现在较低的过饱和度下,在该区间内晶体不生长,该区间的存在首先阻止了体系向平衡态的过渡,降低了产率,其次在多晶型体系下,该区间内可能存在一个长期存在的亚稳相,诱导其他晶型的成核与生长。对于澄清溶液中的均相成核,公式(2)定义了成核的热力学参数B,而非均相成核中,界面能γ应替换为有效界面能γHEN=ψ·γ,从而得到式(3)。对于活性模板,活化因子ψ的值在0~1之间,使B值减小,从而降低了成核势垒。
(2)
(3)
式中:c为形状因子;v为晶体中的分子体积;γ为界面能;k为玻尔兹曼常数;T为温度。
界面能γ受晶核内的分子排列及溶液与晶面表面之间相互作用的影响,因此也可以说热力学参数B包含了成核过程中有关分子水平的信息。CNT将晶核的界面能等同于与同一溶液接触的无限大表面的界面能。通常在分子晶体的成核中,一种结晶化合物的界面能是各向异性的,这也就意味着一个合理的界面能应该是晶核所有表面的加权平均值。Roberts等[20]通过计算阿司匹林晶体界面能,与结晶习性的变化相结合,说明在乙醇水溶液中,随着晶体疏水表面比例的增加,有效界面能从6 mJ/m2到42 mJ/m2不等,这也反映了式(2)和(3)中形状因子c和界面能γ是相互依存的关系,缺少其中任何一个都难以获取另一参数的有效信息。
从动力学的角度来讲,溶液中的团簇自组装之前,生长单元必须首先由一个溶解、溶剂化的状态过渡到一个部分去溶剂化、吸附在晶核表面的状态,扩散过程需要跨越能垒,跨越该能垒需要的活化能与去溶剂化和分子的构象改变有关。式(1)中指前因子A可表达为:
A=zf*C0
(4)
式中:z为Zeldovich因子;f*为分子粘附率;C0为成核点浓度。
根据实验所得成核速率J随过饱和度S的变化曲线,可以得到相应的A值并估计f*C0,但由于除了在一些特殊情况下之外,成核点浓度C0难以确定,因此f*C0是所能获得的有关分子动力学的最有效信息,f*C0可用来推断在不同溶剂中成核分子的相对吸附速率,分子的吸附过程通常与溶液中生长单元的浓度呈线性关系,因此也常用f*C0/M的值来评估不同溶剂中的分子聚集态对成核速率的影响[21],其中M为溶液物质的质量浓度。
Bernstein等提出多晶型体系中,各种晶型是独立的均相成核体系,某种晶型成核和生长是在几种晶型热力学和动力学作用下相互竞争的结果[22-23]。天津科技大学沙作良课题组研究了孕二烯酮在乙醇溶液中的结晶热力学和结晶动力学,得出结论:在低过饱和度下,结晶热力学和动力学都有利于形成稳定晶型,然而在较高过饱和度下,热力学和动力学之间会达到平衡,从而导致产生伴随结晶现象[24]。龚俊波课题组对尿素在水溶液中的成核动力学进行研究,探索了化学势和成核温度对成核速率的影响,虽然化学势差是结晶的驱动力,但它并不总是有利于成核过程。他们证明了当化学势差随成核温度的降低而增大时,动力学因素起主导作用,使成核速率增大;然而,当化学势差随成核温度的升高而减小,在动力学和热力学方面,提高成核温度都有利于成核过程[25]。
1.2 从溶液化学角度理解成核
CNT得出,在溶液中分子团簇克服能量势垒成核和长大。然而,从这种动力学分析,很难得到关于团簇的组成或分子堆积的相关信息。分子在溶液中自组装产生成核前驱体,该前驱体可以直接作为晶体的生长单元,或者经过重排附着到生长团簇上实现成核与生长。Cruz-Cabeza和Davey从溶液化学的角度,结合热力学分析、晶体结构与计算模拟,得到晶核与溶液中生长单元、结构合成子之间的关系,并提出了“成核过渡态”的概念[12]。CNT与非经典成核理论中均未解决晶核中分子堆积方式即晶核结构是什么这一问题[26-27]。
为了从分子水平上理解成核过程,人们结合不同分子可能存在的自组装模式和溶剂化形式开展了大量研究,从溶液化学的角度表征了溶液中分子的存在形态,并与成核过程建立联系。如糖精的晶体结构中存在氢键二聚体[12],如图1所示,对其在不同溶剂中的分析发现,丙酮中分子的主要存在形式为溶剂化的单体,而在乙醇中分子的主要存在形式为溶剂化的单体和二聚体。在丙酮溶液中,成核过程由于涉及单体的堆积错误,最终得到的是孪晶,而在乙醇溶液中,成核过程与二聚体的有序排列有关,得到的是单晶[28]。在甲苯中,2,6-二羟基苯甲酸的存在形态为中心对称的羧酸二聚体,结晶实验中得到的是晶型1[29],而在氯仿中为不具有对称中心的四聚体结构,得到的是晶型2[30],如图2所示。结晶实验与溶液化学的对应结果表明:成核与溶液中分子的自组装过程有关。

图1 糖精晶体结构中的氢键二聚体[12]Fig.1 Hydrogen bonded saccharin dimer extracted from its crystal structure[12]

图2 2,6-二羟基苯甲酸的两种晶型[12]Fig.2 Two polymorphic forms of 2,6-dihydroxybenzoic acid[12]

图3 两种自组装模式下的溶质-溶质分子对(a)氢键相互作用和(b)芳环堆积作用[31]Fig.3 Solute-solute pairs in two self-assembly modes:(a) hydrogen-bonding; (b) aromatic stacking[31]
Cruz-Cabeza和Davey研究了芳香族羧酸苯甲酸(BA)、对甲基苯甲酸(pTA)、对氨基苯甲酸(pABA)和对硝基苯甲酸(pNBA)等分子在不同溶剂中的成核动力学。他们从物理化学的角度,首次考察了分子结构、晶体结构与成核动力学的关系。溶液中这些分子的自组装模式有两种:一种为氢键二聚体(hydrogen-bonded dimers, HBD);另一种为通过芳环堆积作用形成二聚体(stacked dimers, SD),由于芳环上具有不同取代基,且取代基的分子形状不同,由此导致芳环堆积基元的几何排列与重叠程度不同。图3所示为两种自组装模式下分子的几何形态。研究发现相对成核速率并不只是由单体吸附决定,还与对溶剂有依赖作用的芳环堆积相互作用力有关。即晶体的成核速率是由团簇的生长速率所决定的,而团簇的生长是单体分子通过芳环堆积作用附着到晶核表面实现的。芳环堆积作用是成核中的关键步骤[31]。
他们还研究了对氨基苯甲酸(pABA)不同晶型的成核动力学,揭示这两种不同晶型的成核路径以及成核过程溶液中团簇的分子堆积方式和组成。α-pABA中通过氢键相互作用形成了羧酸二聚体,它们沿b轴以平移方式无限堆积,实现了该晶型沿b轴的生长,β-pABA中通过COOH…NH2相互作用形成了层内四聚体结构单元[32],而分子堆积是通过层间形成的二聚体以中心对称方式实现的[33],如图4所示。成核时分子通过芳环堆积作用自组装形成二聚体,在大部分溶剂和过饱和环境下,二聚体中的分子是以平行方式排列的,这种排列方式在几何上有利于后续的分子堆积,即通过酸与酸之间的相互作用形成α晶型。而在特定条件下,如低过饱和度或水溶液中,当二聚体中的分子以反演方式排列时,不同的二聚体之间通过COOH…NH2作用连接形成β晶型,或者在另一种情况下,这种以反演方式堆积形成的二聚体经过重排形成α晶型,图5所示为pABA分子在不同的自组装模式下结晶得到α和β晶型。在这里,水并非提供了溶液介导晶型转变的途径,而是由于pABA和水之间的界面张力较高,使得在某一区间内不发生晶体的成核与生长,α团簇不继续长大,从而为β晶型的出现提供了充足的时间[21,34]。

图4 β-pABA中的分子排列方式[33]Fig.4 β-pABA motifs[33]

图5 pABA的α和β晶型溶液结晶中可能存在的自组装路径[33]Fig.5 Plausible self-assembly pathways in solution crystallization of α and β of pABA[33]
Davey等通过改变溶剂与过饱和度,对大量实验数据进行统计分析,由成核诱导时间得到晶体的成核速率[35]。晶体的相对生长速率越快,达到同等成核速率所需过饱和度的值就越低,说明溶液中由分子到超分子团簇的成核过程,速率限制步骤为团簇生长到临界晶核尺寸[31]。以苯甲酸/甲苯体系为例[35],实验得到的成核速率与过饱和度的关系满足CNT,并利用数值拟合得到成核参数,在分子水平解释了芳香族羧酸在不同溶剂中的成核。

图6 TA晶型Ⅰ的扭转构象和晶型Ⅱ的平面构象[37]Fig.6 Twisted-like and planar-like conformations found in forms Ⅰ and Ⅱ TA[37]
人们还探究了溶液中分子的存在形式对最终生长单元所采取的构象(晶型)是否有影响。以构象多晶型药物托芬那酸(TA)为例,此前Mattei和Li等[36]的研究表明:过饱和度影响TA的分子存在形式,从而影响其晶型。当溶质浓度提高,二聚体数量增多,二聚体中的分子构象与晶型I的扭转构象相同(见图6),从而高过饱和度促进晶型Ⅰ的形成。而Davey等[37]研究发现,TA在甲苯溶液中分子的主要存在形式为HBD,根据Mattei和Li的计算结果,该条件下结晶所得到的应为晶型Ⅰ,而Davey等在同等条件下还得到了TA晶型Ⅱ,说明溶液中分子的存在形式与构象多晶型之间无必然联系,无论分子在溶液中的存在形式为单体或二聚体,最终分子组装成临界晶核所采取的构象不受影响。要想在溶液化学与晶型选择性之间建立联系,应对高过饱和度下亚稳晶型的出现做出解释。乙水杨胺分子在溶液中以一种能量较高的构象存在,这种高能量构象在溶液中的浓度较低,然而在目前得到的乙水杨胺晶体中,乙水杨胺分子并非直接采用这种低浓度的高能量构象,这与分子重排并接触到晶体表面这一过程有关[38]。
溶液中溶剂对溶质分子的溶剂化作用会降低溶质的聚集程度,去溶剂化是形成分子团簇的重要一步。苯甲酸在固态下或在非极性溶剂中都可以形成二聚体结构[39],根据能量计算[21](见表1),苯甲酸在三种溶剂中二聚体的形成倾向由高到低:乙腈>乙酸乙酯>异丙醇,相应的去溶剂化能力也呈现出同样的规律。以苯甲酸-甲苯体系为例,图7(d)为苯甲酸在甲苯中形成的二聚体结构,该二聚体通过与甲苯之间的π-π相互作用被溶剂化,与pABA相比,由于苯甲酸的甲苯溶液中本来就存在二聚体,成核过程不需要首先经历分子的去溶剂化(见图8),因此成核速率更快,在不同溶剂中,pABA的成核速率也随去溶剂化能力呈现相应的变化规律。

表1 pABA在三种溶剂中羧酸二聚体和单体的溶剂化自由能(293 K, 101.325 kPa)[21]Table 1 Solvation free energies of carboxylic acid dimer and two distinct monomers of pABA in three solvents, at 293 K and 101.325 kPa[21]

图7 pABA在(a)异丙醇、(b)乙腈、(c)乙酸乙酯中的溶剂化形式和(d)苯甲酸在甲苯中的溶剂化形式[21]Fig.7 Solvation interactions in solution of pABA in(a) 2-propanol, (b) acetonitrile, (c) ethyl acetate and(d) benzoic acid in toluene[21]

图8 自组装二聚体和溶剂化单体的成核路径[21]Fig.8 Nucleation pathways of self-assembled dimers andsolvated monomers[21]
2 熔体中的经典结晶路径
大部分药物的结晶都是通过溶液结晶实现的,而很少采用熔融结晶。熔融结晶作为一种单一组分体系简化了影响结晶过程的因素,与溶液结晶相比,熔融结晶中无溶剂-溶质相互作用和溶液介导晶型转变。另外,越来越多文献报道,一些难以通过传统的溶液结晶方法得到的晶型,可以在熔融结晶中成功获得,如灰黄霉素(GSF)晶型Ⅱ[40]、ROY晶型Y04[41]、吡罗昔康晶型Ⅵ和Ⅶ[42]。通过熔融结晶筛选得到的新晶型往往比溶液法筛选的晶型具有更高自由能。在固体口服剂型中无定形药物已受到越来越多的关注,因为无定形固体通常可以提高溶解性、溶解速率和低溶性药物的生物利用度。然而,随着时间的推移,高自由能非晶态药物会有再结晶的风险,从而导致治疗效果的丧失。因此,保持无定形药物的物理稳定性对于开发药物配方至关重要。研究无定形药物(熔融)结晶过程具有重要意义。熔融结晶包括晶体的成核和生长,相关研究表明,熔体中的晶体成核速率最大值所对应的温度要低于生长速率最大值所对应的温度,意味着二者有着不同的动力学,需要分开研究。
2.1 熔体中的晶体成核动力学
有机分子熔融结晶的成核动力学报道较少。最近,Yu课题组测量了多元醇(D-山梨醇、D-阿糖醇、D-木糖醇和甘油)[43]在熔体中的成核速率,发现虽然它们结构相似,但是成核速率却相差近10个数量级。这些多元醇的成核速率可以通过CNT拟合出来,说明它们成核过程符合经典的成核理论。根据CNT[44-46],熔融状态下的晶体成核速率可表示为:
J=kJexp(-WC/kT)
(5)
(6)
式中:WC为成核的热力学势垒;σ为晶核与熔体界面自由能,需要指出的是,界面能σ微小差别就可以导致成核速率数量级差别,因此,在利用CNT解释晶体的成核过程时,应尽可能得到成核界面能σ的准确值;ΔGV为晶体与熔体自由能之差;kJ为动力学因子,表示分子与晶核结合的频率,可以表示为一些熔体动力学参数的函数,如式(7)~(9)所示。
kJ=fDD
(7)
kJ=fη/η
(8)
kJ=fτ/τα
(9)
式中:D为扩散系数;η为黏度;τα为结构弛豫时间;fD,fη,fτ均为温度敏感性常数。
Yu课题组发现,用晶体的生长速率u可更加简单且准确地计算出成核动力学因子[43]:
kJ=fuu
(10)
式中:fu为温度敏感性常数。
最近,Cai课题组研究了三种聚合物(HPMCAS、PVP、PEO)对氟康唑无定形成核速率的影响[47],发现HPMCAS能够明显抑制氟康唑无定形成核速率,PVP对其成核速率影响不大,而PEO则能明显加快其成核速率。该研究为如何选择稳定无定形聚合物提供了指导。
熔融结晶过程中可能会发生交叉成核现象,Yu课题组提出了一种新的成核机理:交叉成核(cross nucleation)[48-49],即最先成核的晶型(A)不会消耗完所有溶质或发生晶型转变,而是作为模板使另一种晶型(B)成核,相较于原始晶型(A),新晶型(B)的热力学稳定性可能更高也可能更低,生长速率大于等于原始晶型(A)。交叉成核与溶液介导相变中的二次成核有一定联系但又存在差异[50-52]:相同的是,二者都是界面成核;不同的是,溶液介导相变中,二次成核的晶型一定比初始晶型有更高的热力学稳定性。需要指出的是,交叉成核并非晶型转变,D-甘露醇的α晶型在δ晶型上交叉成核证实了这一点[52]:(1)尽管δ晶型的稳定性低于α晶型,但在实验条件下δ晶型有足够的稳定性,并且实验过程中该晶型仍然在生长;(2)晶型转变速率应随温度的升高而加快,但α晶型在δ晶型上出现的速率随温度升高而减慢,并且实验中观察到α晶型也可以在D-甘露醇的最稳定晶型β上成核。
2.2 熔体中的晶体生长动力学
在熔点附近,晶体生长速率是由热力学控制,温度降低,热力学驱动力增加,晶体生长速率加快;进一步降温,晶体生长速率由分子扩散限制,扩散速率随温度降低而减慢,最终导致生长速率减慢;当温度降低至Tg附近,生长速率异常加快,比分子扩散模式下的生长速率高出几个数量级,这种新的晶体生长模型被称为“玻璃态-晶体生长模型(glass-crystal growth mode)”,简称为GC生长模型,这种快速生长模式不仅仅出现在Tg以下,在温度上升至1.15Tg时的熔融态下仍存在[53-55]。一些理论认为,GC生长模型可归因于均相成核[56]、张力诱发分子迁移[57]、由局部分子迁移引发的固态相变[53]等。对于储存温度在Tg以下的无定形药物,研究GC生长模型对其稳定性研究具有重要意义。
熔体中的晶体生长另外一个重要特征是熔体表面的晶体生长速率要远大于熔体内部的晶体生长速率,这是因为熔体表面分子迁移率要远高于熔体内部的分子迁移率,如熔融态的硝苯地平[58]和灰黄霉素[59]的晶体表面生长速率均远高于内部。上文提到,一些有机小分子在Tg附近具有GC生长现象,虽然GC生长是一个内部过程,但它可能与表面分子迁移率存在一定关系,这是因为晶体本身的密度高于玻璃态,晶体生长过程中的最前端会不断产生微裂纹(自由表面),使分子有了较高的迁移速率而加快生长速率。对于不同体系来说,表面扩散速率存在较大差异,造成这种差异主要有两方面原因:一方面是分子尺寸,即表面分子扩散速率随分子尺寸增大而减慢;另一方面是分子间氢键,即表面分子扩散速率随分子间氢键增多而减慢。例如,灰黄霉素仅有氢键受体而无氢键给体,因此其晶体结构中没有分子间氢键,该体系下表面分子扩散速率不受氢键影响,并符合“无分子间氢键”的无定形分子表面扩散速率的总体趋势,即表面扩散速率随分子尺寸的增大而减慢。为了解决无定形药物表面分子迁移率较高使药物稳定性降低的问题,目前常采用纳米包覆的方法提高稳定性,如将无定形吲哚美辛用聚二烯丙基二甲基氯化铵(PDDA)[60]、明胶[61]或壳聚糖[62]包覆后,其稳定性明显改善。另外也可采用引入聚合物添加剂抑制无定形药物的成核与生长过程,如聚乙烯吡咯烷酮(PVP)的加入提高无定形D-山梨醇和D-阿糖醇的稳定性[63]。
3 溶液中的非经典结晶路径
随着对成核研究的深入,由于CNT模型过于简化,它的不足之处也被人们发现。在蛋白质结晶的成核动力学研究中,Vekilov课题组发现,实验中测得的成核速率比根据CNT的预测值要低十个甚至十几个数量级[64-65],他们通过动态光散射技术,证明在过饱和溶液中,会先形成蛋白质分子的致密无序团簇,随后从该致密无序团簇中形成晶核,并提出了两步成核机理[66]。Myerson课题组用近红外激光对甘氨酸溶液进行辐照,采用线偏振和圆偏振能分别得到甘氨酸的不同晶型(γ和α),这个结果可以证实甘氨酸的两步成核机理,即分子聚集形成晶核前会经历一个高密度中间态[67]。后来他们利用小角X射线散射(SAXS)证实了甘氨酸的两步成核过程,即单分子—二聚体—晶核这一成核路径[68]。近十几年,一系列原位表征技术的崛起(冷冻电镜、原位透射电镜、原位原子力显微镜)使在分子以及纳米尺度直接观察成核过程成为可能[69-71]。根据实验观察,除了经典成核路径,经历多核团簇、亚稳晶态、无定形或致密液态等中间态的多步成核路径被提出[9-11],即非经典成核路径(non-classical nucleation pathway)。研究者们利用原位表征技术不仅仅对蛋白质晶体的成核与生长,对生物矿化[72-73]、硅沸石[74]、无机纳米材料[75-76]和有机晶体[77-78]等材料的成核和生长过程也进行了原位观测,并对其非经典结晶路径进行报道。
与经典结晶路径不同,非经典结晶中采用的生长单元不再是原子、离子或分子,而是更大的前驱体粒子[9,79],涉及成核前团簇(pre-nucleation cluster, PNC)的形成和两步成核机理[11]。De Yoreo提出非经典的晶体生长机制中存在粒子在特定晶面上的定向附着(oriented attachment, OA),OA使一些纳米晶粒、低结晶度的纳米晶粒生长为单晶[9],另外,最近的研究表明,OA还促成了一种重要的孪晶结构——五重孪晶的形成[80],这种OA所导致的晶体生长受粒子间界面力的影响[81-82]。许多物质都采用了非经典的结晶路径,如蛋白质的结晶经历了由凝聚态液相到有序晶体转变的过程[64-66,83],碳酸钙的成核也与溶液中成核前团簇的形成有关[72,84-86]。药物结晶中前驱体的存在会影响最终产物的晶型,无论前驱体为非晶态或晶态,或由短暂存在的亚稳态组成,对其识别并进一步表征将反映最终稳定存在晶体的有效信息,对于多晶型体系来说尤其重要。
Myerson课题组研究发现激光可以诱导甘氨酸过饱和溶液的成核,并达到较快的成核速率[87],用同样的方法,电子束与溶剂相互作用产生化学辐射可诱导氟芬那酸(FFA)的成核[88]。FFA的结晶溶液中存在3 nm和8 nm的粒子,其中不同数量的8 nm粒子会聚集到一起形成新的凝聚态团簇,根据聚集程度的不同将这些凝聚态团簇划分为三个类别,而之所以认为这些小颗粒是非经典成核中的PNCs,是因为他们导致了新的亚稳相的形成。如图9所示,FFA非经典结晶的中间过程可总结为以下两点:(1)PNCs聚集到一起,界面消失,结合成亚稳的高密度液相凝聚态(dense liquid phase, DLP),如图10所示;(2)凝聚态的密度提高,趋向于晶态,并不断吸收周围溶质,相对于亚稳态前驱体密度提高,如图11所示。其中PNCs是结构有序的结晶态,因此在溶液中可以稳定存在。

图9 FFA非经典结晶过程:聚集、凝结、致密中间体的成核、结晶[88]Fig.9 Nonclassical crystallization process of FFA: aggregation, coalescence, nucleus formation from the densifiedintermediate material and crystallization[88]

图10 PNCs随时间变化的聚集形态,伴随着界面的消失,PNCs聚集形成一个连续的凝聚态需要180 s(标尺:20 nm)[88]Fig.10 A time-lapse of each of the aggregates of PNCsundergoing coalescence. The process for the aggregated PNCs to coalesce and create a cohesive substance took 180 s, where separation between PNCs gradually reduced until there was no observable boundary between PNCs (Scale bars: 20 nm)[88]

图11 FFA结晶前三种凝聚态团簇的形貌变化过程:高密度核形成,然后消耗亚稳连续相中的物质,直到最终形成一个高密度的整体(标尺:20 nm)[88]Fig.11 Morphological changes of the three aggregates illustrating the pre-crystallization process of FFA: an electron dense nucleus develops and then consumes the material in the metastable cohesive entity until a final electron dense entity was formed (Scale bars: 20 nm)[88]
Cölfen等[89]用电位滴定法监测了布洛芬成核的早期阶段,布洛芬成核前经历了液-液相分离过程,形成DLP,并以此作为成核的前驱体,如图12所示。2D1H-1H NOESY NMR研究表明,DLP中和晶体中的分子间距离是类似的,在DLP内,与分子平移和旋转相关的分子动力学过程减慢,布洛芬分子浓度更大,黏度高于周围的液体环境,这也解释了DLP在成核前一段时间内可以稳定存在的原因。Vekilov等[64]也提出黏度对DLP中间体内有序核的形成起关键作用。
在过饱和的L-谷氨酸水溶液中,由于溶液与非晶态液滴之间的界面能较低,在能量上有利于形成非晶态中间体,如图13所示,这种非晶态中间体是成核前团簇聚集形成的,随后,非晶态中间体进一步合并,体系的总表面积减小,晶体的成核发生在非晶态中间体的内部,形成了两步成核过程[90]。非晶态中间体的存在不仅仅决定了初始成核过程,也对晶体形态、尺寸分布、平均尺寸和相变过程产生影响。Cölfen课题组[77]采用电喷雾电离质谱(ESI-MS)、分析超速离心实验(AUC)和原位原子力显微镜(in situ AFM)等表征手段,研究了DL-谷氨酸一水合物表面及溶液中存在的前驱体团簇,如图14所示为团簇存在时晶体的多步结晶路径:溶液中成核前团簇发生相分离,然后聚集形成更大的纳米液滴,液滴吸附到晶体表面并扩展使界面能降低,三维纳米前驱体逐渐转变为二维晶核,在溶液中纳米液滴的补给下,二维晶核最终长成一个新的分子层。由此可见,这种无序的纳米前驱体不仅仅与晶体的成核过程有关,在生长后期还起到了物质储存的作用。

图12 布洛芬的水解平衡及成核和晶体生长路径[89]Fig.12 Protolysis equilibrium of ibuprofen with pathway of nucleation and crystal growth[89]

图13 L-谷氨酸亚稳区(MSZ)相分离初期非晶态中间体的TEM和SAED图像,(a1~a4)、(b1~b4)和(c1~c4)分别对应不同的亚稳区间[90]Fig.13 TEM images and SAED pattern of amorphous intermediates in initial stages of phase separation of L-glutamic acid in the metastable zones (MSZ): (a1~a4),(b1~b4) and (c1~c4) correspond to different MSZ respectively[90]
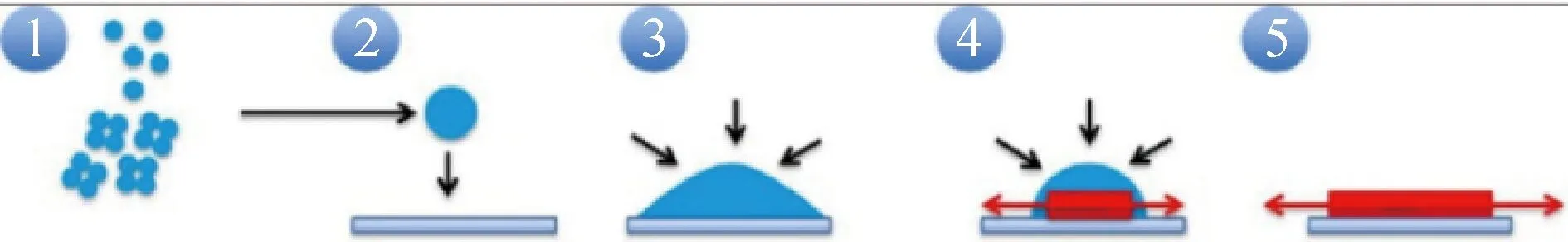
图14 DL-谷氨酸一水合物晶体的多步生长路径[77]Fig.14 Multistage growth pathway of DL-glutamic acid monohydrate crystal[77]
奥氮平(OZPN)是一种抗精神病药物,属于BCS Ⅱ类药[91],目前所得到的该药物所有晶型(OZPN I, OZPN Ⅱ, OZPN Ⅲ)和三种二水合物(OZPN DB,OZPN DD,OZPN DE)中分子均以二聚体形式排列。Florence课题组[92]研究表明,在93%的相对湿度下,OZPN晶型I的表面可得到OZPN DD,而在搅动的水溶液中,晶型I表面首先产生OZPN DB,随后转变为更稳定的OZPN DD。OZPN I表面OZPN DD的产生可总结为以下几步:如图15所示,A密集的纳米液滴在OZPN I (100)面的台阶处形成,B液滴进一步聚集,C形成具有晶体特征的介观尺度的液滴,D完全形成OZPN DD晶体排列在OZPN I (100)面上。OZPN晶型I在这里充当了模板的作用。当溶液处于搅动条件下,液滴从晶体表面脱附,失去模板效应,成核过程遵循路径E~G,产生OZPN DB。这些密集的纳米液滴不是一种稳定相,它们沉积在晶体表面后通过与其他团簇凝结实现生长,即使在不饱和的OZPN水溶液或水-乙醇溶液中也存在,它们本质上是无序的,平均半径为35 nm,在水溶液中,AFM下观察到OZPN晶体(100)面上吸附着大小相近的团簇,如图16(a)所示。非晶态团簇尺寸不随OZPN浓度和溶剂组成而改变,根据模型假设OZPN分子形成二聚体并供给团簇生长,可以推断:这种瞬态二聚体结构与OZPN已知晶体结构中的中心对称二聚体相似,并且作为团簇基本组成单元存在[93]。

图15 OZPN DD在晶型I表面和OZPN DB在溶液中的成核过程[92]Fig.15 Nucleation process of OZPN DD on the surface of OZPN I crystal and OZPN DB in solution[92]

图16 AFM下OZPN凝聚态团簇(箭头所示)在晶体表面的吸附 (a)在水中4 h后OZPN晶型Ⅰ(100)面的团簇,作为无水OZPN晶型I转变为水合物OZPN DD的动力学中间体;(b)EtOH/H2O 1/1(v/v)饱和溶液中1 h后OZPN/EtOH/H2O表面的团簇[93]Fig.16 Dense OZPN-rich clusters (indicated with arrows) on the surface of OZPN crystals imaged by AFM(a) clusters on (100) face of OZPN I after incubation in water for 4 h, these clusters are a kinetic intermediate in the transformation from anhydrous OZPN I to the hydrated form OZPN DD; (b) clusters on the surface of OZPN/EtOH/H2Oafter incubation for 1 h in saturated solution in EtOH/H2O 1/1 (v/v)[93]
晶体生长中生长单元到达台阶上的扭结点处具有两种方式,生长单元直接结合到扭结点处或生长单元首先吸附到台阶面上,然后扩散到扭结点,如图17所示,经典的晶体生长理论认为,晶体表面台阶的生长是通过溶质单体的结合实现的[94],这种单分子反应使得台阶生长速率v与溶质浓度C之间呈线性关系,即:
v=βΩ(C-Ce)
(11)
式中:Ce为溶解度,说明分子吸附的可逆性;Ω为晶体内的分子体积;β为有效动力学系数,其中包含了与生长机理有关的动力学参数。
Vekilov课题组[95]对公式(11)作了进一步的推导,得出溶液中存在二聚体时台阶速率与溶质浓度的关系,如表2所示,所对应的曲线如图18所示。奥氮平的2OZPN·EtOH·2H2O晶体台阶生长速率v与溶质浓度C之间的关系满足上述第二种情况,生长速率v与溶质浓度C的二次方成正比,如图19所示,拉曼测试与模拟进一步表明:晶体生长是以与溶质单体共存的二聚体为生长单元实现的。AFM下观察到,具有较宽台阶面的台阶生长速率较快,证明了表面扩散机制的存在。

图17 溶液中的生长单元结合到扭结点处的两种方式(i)直接结合; (ii)首先吸附到台阶面上,然后向台阶扩散[95]Fig.17 Two pathways of a growth unit from solution to kinks: (i) direct incorporation and (ii) via adsorption on the terraces followed bydiffusion towards the steps[95]

图18 不同情况下台阶速率与溶质浓度的关系(分别对应表2中的四种情况)[95]Fig.18 Correlations between step velocity and soluteconcentration under four cases (corresponding to four cases in table 2)[95]
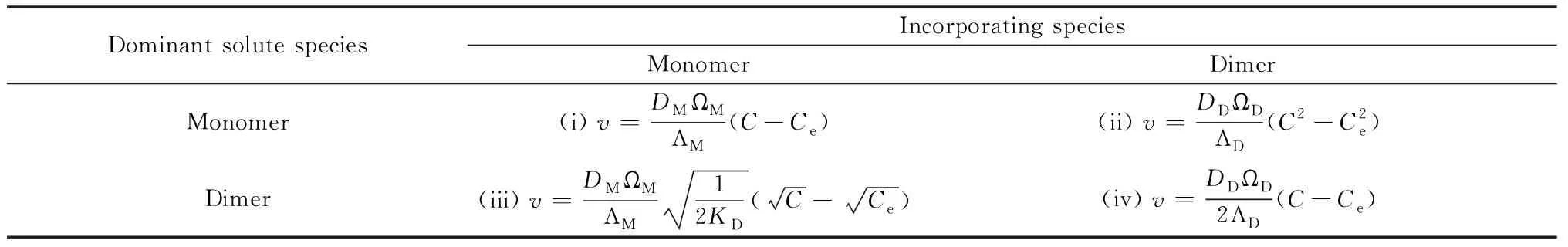
表2 根据溶液中主要组分及生长单元类型划分的四种情况下台阶速率v与溶质浓度C之间的动力学方程[95]Table 2 Kinetic laws that correlate step velocity v to the total solute concentration C for four cases classified according to the dominant solute species in solution and the form that incorporates into the steps[95]

图19 2OZPN·EtOH·2H2O晶体生长台阶生长速率v与溶质浓度C之间的关系[95] Fig.19 Correlations between step velocity v and solute concentration C of 2OZPN·EtOH·2H2O crystal[95]
4 结语与展望
由于药物分子的性质及生物利用度与其晶型息息相关,而最终产品的晶型由其成核和生长路径决定,因此,对于药物分子,其成核和生长路径研究就变得非常重要,其结晶过程中是否经过中间态?或者是否经过瞬时的亚稳晶型转化而来?这些中间态与其最终晶型有怎样的联系?了解这些对于进行晶型主动调控,获得具有自主知识产权的优势晶型有着重要的意义。从溶液化学的角度,根据晶核或PNC中的分子堆积方式与体块晶体结构合成子是否存在对应关系,可以进一步解释经典结晶与非经典结晶路径。一方面,当晶核或PNC中的分子堆积方式与晶体结构合成子之间存在直接对应关系时,此时CNT是适用的;另一方面,当二者不存在一一对应关系时,溶液中已经形成的分子团簇可能充当了分子重排的位点,从而产生最终晶体结构中的超分子合成子,这种两步自组装过程对应了非经典的结晶路径。
对于一个给定的药物分子,可以通过分析分子结构和化学性质判断可能存在的分子自组装模式,结合计算模型进行稳定性分析,得到分子的稳定存在形式,即得到溶液化学的有效信息。即便如此,也很难预知分子在参与晶体生长时的复杂运动过程,如分子重排等。非经典结晶路径涉及的中间态前驱体团簇通常也是难以预知的,需借助先进表征手段。因此,仅仅通过分子结构和化学性质难以预测药物晶体的结晶路径。尽管对非经典结晶过程的表征困难重重,而且目前还缺乏可靠的理论体系来辅助解释该结晶过程,对该路径的研究仍是极其必要的。从分子水平研究溶液、熔体中成核和生长过程,有赖于先进实验手段的采用和理论的发展,药物晶型控制是一个即具有基础研究意义又具有巨大应用前景的领域,值得更多的晶体生长工作者关注!
猜你喜欢
学与玩(2022年12期)2023-01-11
化工学报(2022年7期)2022-08-10
弹性体(2021年6期)2021-02-12
陶瓷学报(2020年2期)2020-10-27
上海大学学报(自然科学版)(2020年2期)2020-05-13
陶瓷学报(2019年6期)2019-10-27
中国塑料(2016年9期)2016-06-13
中国塑料(2015年6期)2015-11-13
中国塑料(2015年8期)2015-10-14
中国塑料(2015年7期)2015-10-14
