
罕见46,XY/47,XYY嵌合体型女性患者病因诊断的遗传咨询模式探讨
2021-05-31 02:43孙艳王桂琪杨文惠莫中福
国际生殖健康/计划生育杂志 2021年3期
孙艳,王桂琪,杨文惠,莫中福
性反转综合征(sex reversal syndrome)是一类性别发育异常的人类遗传学疾病,其主要特征是染色体性别与性腺性别以及表型性别不一致,疾病类型主要包括46,XX男性性反转和46,XY女性性反转。目前已经发现了很多参与性别决定或者睾丸发育决定的基因,大多数以性别决定基因(SRY基因)为主导,极少数情况下也存在非SRY基因主导的男性性别发育。由于这些基因调控的过程较为复杂[1],因此任一环节的误差都可能造成多种多样的性别异常。在临床遗传咨询中优化诊断路径更快更准找出遗传病因和全面了解性反转的致病机制极为重要。46,XY性反转综合征又称为Swyer综合征,近几年在临床较为多发。从致病基因分析发现,仅有10%~15%左右Swyer综合征病因为SRY基因突变或缺失所致,而80%以上外生殖器性别不明合并生殖腺发育不全的XY女性病因不明[2-3]。本文报告1例罕见的46,XY/47,XYY嵌合体女性性反转病例,并基于染色体核型分析、染色体微重复微缺失拷贝数变异测序(copy number variation-sequencing,CNV-seq)分析以及家系和临床表型分析,探讨46,XY女性性反转发生的可能遗传学机制,并基于这种分析模式,提供快速指向性的基因检测咨询建议,从而优化此类性反转患者的遗传诊断路径。
1 资料与方法
1.1病例资料患者,23岁,社会性别为女性,青春期后月经一直未来潮,体格检查:身高165 cm,可见乳房发育,略有喉结,有体毛、腋毛等。外生殖器异常,阴毛呈女性化分布,阴蒂长约1.4 cm,前端似龟头状,无孔,双侧大阴唇肥大,质软,无压痛。小阴唇小,有尿道孔,无阴道。双侧腹股沟有1.8 cm×0.9 cm的包块。B超示:右侧髂脊内侧可见1.2 cm×0.6 cm的低弱回声区,盆腔、腹腔均未探及到子宫和卵巢,会阴部未探及明显睾丸样声像图。腹腔镜检查见:盆底部可见条索状始基子宫,双侧附件区均可见似输卵管样结构,左侧输卵管下方可见卵巢样组织,大小约为1.0 cm×0.6 cm×0.5 cm,色白,质地偏韧,取左侧卵巢样组织及腹股沟包块行病理活检。否认家族史。实验室检查:雌二醇20.1 pmol/L,卵泡刺激素45.14 mIU/mL,泌乳素323.7 mIU/mL,孕酮0.503 mIU/mL,睾酮1.104 mIU/mL,可见雌激素水平低下,卵泡激素水平升高,睾酮水平稍高于正常女性。
1.2方法取外周血淋巴细胞培养,常规方法收获细胞、制片、G显带,莱卡染色体扫描分析仪进行染色体核型分析,此病例特殊,特此计数108个中期分裂相细胞,分析10个核型。
采用高通量测序对23对染色体进行CNV-seq检测,机器型号:华大BGI500,所用检测试剂均由华大提供,实验步骤均严格按照实验室的标准操作流程(Standard Operation Procedure)文件进行操作。
2 结果
2.1诊断该例女性患者的外周血染色体核型为:46,XY[57%]/47,XYY[13%],见图1。高通量测序染色体检测示:Arr [hg19]46,XY [70%]/47,XYY [30%],Yp11.31-q11.223×3,dup(6)(q14.1)。病理学检测结果:左侧为类似卵巢皮质结构的纤维组织,腹股沟包块为睾丸组织,综合考虑确诊该患者为46,XY/47,XYY嵌合体型女性性反转综合征。高通量测序检测结果见图2,6号染色体长臂的重复所涉及PHIP基因与炎症相关,IRAK1BP1是糖尿病相关基因,与本文性反转综合征无相关性。

图1 外周血淋巴细胞染色体核型分析

图2 外周血拷贝数变异检测结果
2.2遗传家系分析患者为先证者,同胞有一哥哥和一妹妹。哥哥和妹妹均有正常生育史。后经家系调查发现,其母系亲属中还存在与先证者类似遗传表型的个体,分别是其姨外祖母(外婆的姐姐)、一位姨妈(妈妈的妹妹)和一位姨侄女(先证者妹妹的女儿,2岁),而先证者父系家族中均无类似病例,家系图谱见图3。先证者父亲查体:性腺发育与其他性征完全正常,外周血淋巴细胞染色体制备G显带分析其核型为46,XY;AZF基因所检测位点均未见缺失,SRY基因检测阳性。其母亲查体:乳房发育正常,外阴阴毛较少,大小阴唇正常;外周血G显带核型为46,XX;AZF基因及SRY基因检测均为阴性;婚后1年足月顺产一男性婴儿,一切正常;先证者为其母第二胎。本研究家系调查发现(家族亲属描述),所有家族成员中,男性均为正常表型和生育能力,而性征异常女性患者均无生育史,并且性征异常女性成员中,除了具备女性外生殖器官之外,均有类男性外阴的发育不全器官遗迹,原发性闭经。同时检测该家系其他患者核型及拷贝数变异并对Y染色体上的SRY基因和AZF基因位点进行检测。结果示:先证者母亲和妹妹核型均为46,XX;拷贝数变异检测同样提示Arr[hg19]46,XX[100%],未发现其他明确致病性拷贝数变异(CNVs);SRY基因检测阴性,AZF基因所检测位点均未见缺失。

图3 遗传家系图谱
2.3病因诊断的遗传咨询模式对46,XY女性性反转病因诊断提出一种快速诊断路径,见图4。
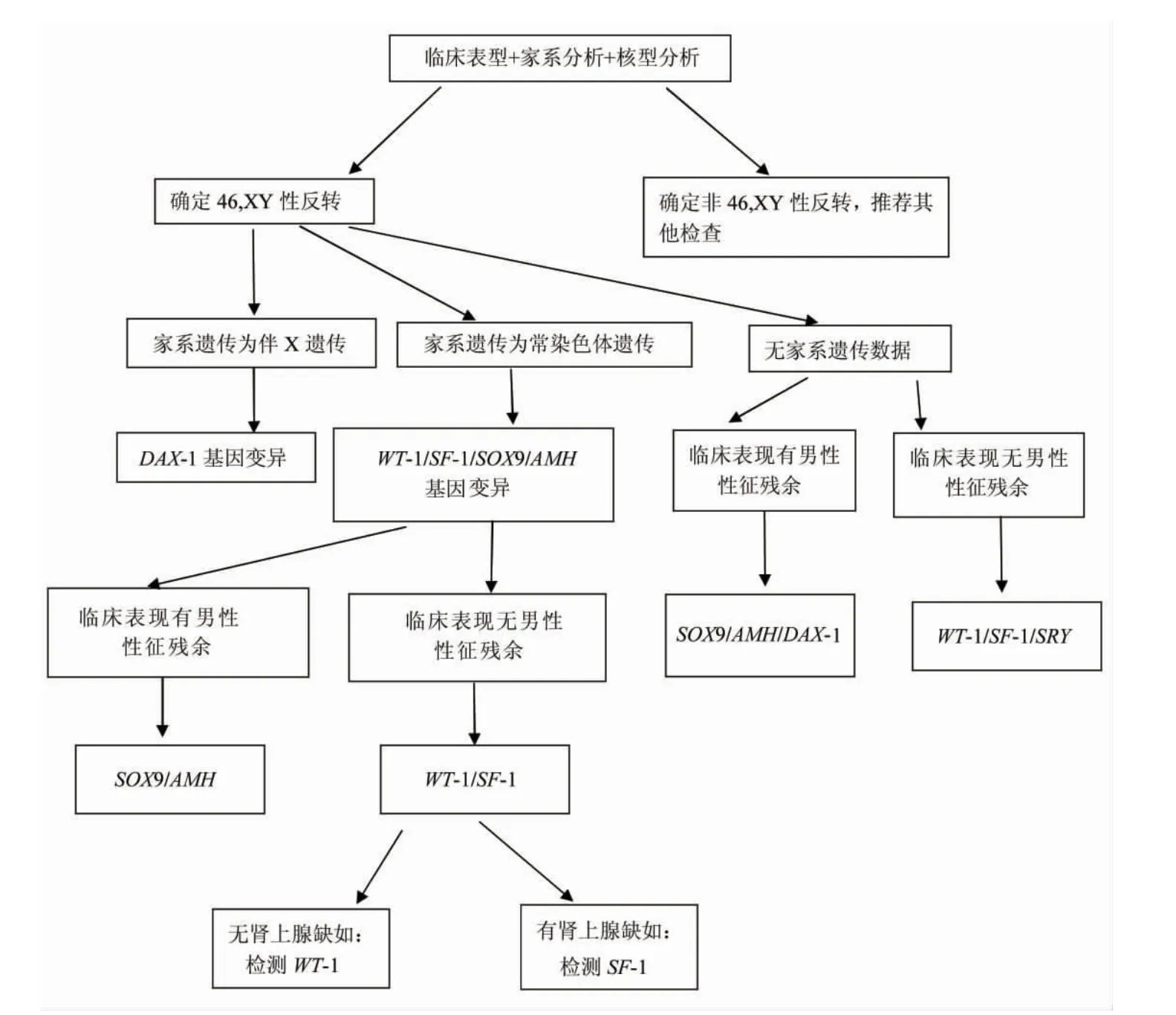
图4 46,XY女性性反转病因快速诊断路径的遗传咨询模式图
3 讨论
3.1SRY基因及其遗传控制模式与临床表现人类性别的决定除了由位于Y染色体上的SRY基因控制之外,还有许多位于其他染色体上的性别决定相关基因控制这一发育过程。在其他性别相关基因的调控表达正常情况下,SRY基因的突变与缺失基本构成了46,XY女性性反转的主要遗传机制,但是这一原因所占比例仅为10%~15%,其他80%以上的病因均为非SRY基因异常所致。目前已知的非SRY基因异常所致46,XY性反转涉及到的基因主要包括如下5类。
3.1.1 WT-1基因 人类的WT-1定位于11p13,WT-1突变可引起Denys-Drash综合征和Frasier综合征,患者同时伴有性反转现象和生殖器发育异常。缺乏WT-1(+KTS)的小鼠,会引起Sry表达水平降低,引起XY小鼠发生完全性反转,研究发现,WT-1(+KTS)基因对维持SRY基因的正常表达水平具有决定作用,对维持男性性征发育具有决定作用。因为位于11号染色体,该基因的性状控制遗传模式为常染色体隐性遗传。
3.1.2 类固醇生成因子-1(steroidogenic factor-1,SF-1)基因 人类的SF-1基因定位于9q33。在XY核型性腺中的支持细胞、睾丸间质细胞中,都有SF-1的表达。SF-1通过与WT-1协同作用,促进抗苗勒管激素(anti-Müllerian hormone,AMH)在支持细胞中的表达,从而诱导和维持雄性正常发育。临床研究发现,无论什么染色体核型的个体,一旦SF-1功能障碍,外生殖器均表现为雌性。并且人类的SF-1突变会导致先天性性腺发育不全和肾上腺缺如及伴有性反转等症状。由于SF-1为WT的协同作用因子,并且位于9号常染色体上,因此,其遗传控制模式同样为常染色体隐性遗传。
3.1.3 SOX9基因 该基因与SRY有较高同源性,目前在人类及小鼠中已发现其家族成员有三十余个,其中SOX9对性别决定尤为重要。人类的SOX9基因定位于17q24.3~25.1。SOX9的表达与支持细胞的分化密切相关,其作用主要是对SRY基因的调控功能起协同作用。SOX9基因突变会导致XY核型患者表现为女性或者双性体。该基因的遗传模式同样为常染色体隐性遗传。
3.1.4 DAX-1 基因 人类DAX-1 基因定位于Xp21.3~21.2,DAX-1蛋白,其表达蛋白缺少典型的锌指结构,不直接与DNA结合,其在基因网络中的调控模式是其蛋白产物间的相互作用调控,因此,该调控模式与基因表达剂量呈现密切的相关性。在正常情况下,DAX-1为SRY所抑制,当其有2个重复的活性拷贝时,可导致SRY正常的XY个体发育为女性。因此,DAX-1与SRY基因在性别决定的平衡中存在竞争抑制关系,二者在剂量上的差异,决定了患者个体的两性性征表现程度。譬如本例先证者,由于存在XYY嵌合体,SRY的剂量更高于一般XY核型个体,因此在性反转女性体内,男性性腺与外生殖器的表现度更显著。由于DAX-1基因伴X遗传,因此该基因导致的性反转性状的遗传模式为伴X染色体不完全隐性遗传。
3.1.5 AMH基因 AMH基因位于人类染色体的19p13.3~13.2,是雄性性别分化的重要因子,AMH的表达使Müller管退化,从而抑制雌性生殖管道发生。SF-1和SOX9是AMH的上游刺激表达调控元件,在调控通路上共同为SRY表达以及雄性发育起到促进作用[4]。
以上SF-1与WT-1在未分化性腺的发育早期出现,因此此类基因的变异将导致性反转患者的雄性性征全部缺失;而SOX9和AMH等基因的表达处于较晚发育时期,因此,基因突变患者往往会出现部分雄性生殖腺体或器官残余。而DAX-1是唯一直接参与雌性性别决定过程的基因,对雄性发育过程通过剂量效应实施不同等级的抑制程度,2个拷贝有活性的DAX-1表达就会引起XY个体发生性反转,但是剂量不足时会导致雄性性征残余甚至双性出现[5]。
3.2本例患者遗传机制与致病基因的分析本研究先证者在临床表现上,出现雄性性征残余(隐睾组织、喉结、体毛较重、睾酮异常升高等),因此从发育过程推测,患者遗传病因可能为性腺分化较晚时期相关基因或者雄性抑制基因突变所致,所涉及到的基因可能有SOX9、AMH和DAX-1。从文献来看,SRY基因突变或者缺失案例中,患者均表现完全的女性性征发育不全,而没有任何男性性征残余[6]。因此,本案先证者的病因不可能是SRY基因变异所致。
通过细胞遗传学和分子遗传学检测验证,发现先证者除了具备XY核型之外,还有约30%的XYY嵌合体状态。理论上,从DAX-1基因与SRY基因遗传的剂量平衡状态分析,具有2个拷贝SRY多剂量的嵌合状态个体,在临床表型上将具有更多的男性性征出现。先证者的临床表现与遗传学鉴定结果高度吻合。与先证者及家属通过多方面沟通,家属一再表示暂时无再生育要求拒绝全外显子测序验证DAX-1基因这一致病位点。
进一步分析:从SRY基因在本研究先证者的临床表现度来看,46,XY/47,XYY嵌合体型女性性反转的性征差异,介于单纯的46,XY女性和47,XXY克氏综合征患者之间。克氏综合征也即先天性睾丸发育不全,患者在儿童期无异常,常于青春期或成年期出现异常,患者男性第二性征发育不完全,外生殖器和生殖腺发育不足——这一点与本例先证者以女性性征外观为主不同。由此推测,本例先证者的DAX-1变异导致的获得性功能,远远超过克氏综合征中仅仅由DAX-1剂量加倍带来的对SRY抑制竞争效应。故初步推断,本例DAX-1基因可能是获得了超强表达变异,或者因为位点突变获得了更强烈的SRY结合特性,导致SRY功能被严重抑制,呈现类似基因沉默的效应。下一步我们将着重研究揭示DAX-1基因变异的遗传学特征与生物学功能。
3.346,XY女性性反转诊治体会46,XY女性性反转患者通过结合临床表现、家系遗传模式分析以及细胞遗传学乃至CNV-seq高通量测序检测,快速确定患者在基因变异病因上的可能性,从而为患者提供下一步进行经济快速的相关基因检测项目提供最可靠的依据。
因此,最终将该患者的遗传病因高度怀疑为母系X染色体上DAX-1变异所致,并据此形成遗传咨询意见,为患者下一步遗传病因确诊提供明确的检测指导。家系已有2岁的患者出生,如有再生育要求应行产前诊断,对先证者及先证者母亲及家系中所有患者和携带者DAX-1基因进行检测以进一步明确此家系的致病基因位点,一旦明确致病基因位点如选择再生育则采用胚胎植入前基因诊断(preimplantation genetic diagnosis,PGD)选取正常的胚胎,避免此类患儿的出生。
猜你喜欢
临床输血与检验(2022年3期)2022-06-22
宁夏医学杂志(2020年3期)2021-01-21
郑州大学学报(医学版)(2019年3期)2019-06-03
传染病信息(2019年2期)2019-05-17
科学之谜(2019年3期)2019-03-28
科学之谜(2018年8期)2018-09-29
恋爱婚姻家庭·养生版(2016年9期)2016-09-07
哈尔滨医药(2015年2期)2015-12-01
中央民族大学学报(自然科学版)(2015年2期)2015-06-09
重庆医学(2015年12期)2015-03-05
