
K2ZrF6 对镁合金微弧氧化膜抗点燃性能的影响
2021-07-03 09:25韩东连勇张津
表面技术 2021年6期
韩东,连勇,张津
(1.北京科技大学 新材料技术研究院,北京 100083;2.北京市腐蚀/磨蚀与表面技术重点实验室,北京 100083)
镁合金因密度低、比强度和比刚度高以及良好的阻尼性能和电磁屏蔽性能等,成为极富潜力的轻量化材料,在航空航天、轨道车辆、汽车工业等交通领域具有广阔的应用前景[1-2]。由于镁元素金属活泼性较高,氧化膜疏松、不够致密,在高温下具有较高饱和蒸气压,增加了小尺寸薄壁镁合金零件在火灾或与其他热源(如电火花、冲击碰撞)接触时发生起火燃烧的风险[3]。而且,镁的燃烧热值高,燃烧速度极快,一旦燃烧,会在极短时间内释放更多的热量。更为严重的是,目前交通工具配备的灭火器难以扑灭镁合金燃烧产生的火焰,因此会导致灾难性的后果[4]。
为降低镁合金起火燃烧的风险,目前主要采用合金化的方法,向镁合金中添加Be、Ca 或者稀土元素来提高镁合金的起燃温度[5-6]。但是,添加的合金化元素只有达到一定含量后才能起到阻燃作用,且如果这类合金化元素含量过高,往往会降低镁合金的力学性能,同时也增加了制备工艺的复杂性和镁合金的成本[7]。
采用表面处理是进一步提高镁合金抗点燃性能的重要方法。目前,利用微弧氧化技术对镁合金进行处理的主要目的是提高其耐腐蚀性能或耐磨损性能,未见提高其抗点燃性能的报道[8],而微弧氧化涂层在提高铝、钛等轻质合金热防护方面已经取得一些应用进展。王亚明团队[9]用氧乙炔焰对铝合金微弧氧化层进行了烧蚀测试,结果表明,微弧氧化层有良好的耐冲刷、抗火焰烧蚀性能,铝合金基体无变形,而无微弧氧化层的基体铝合金变形严重。某型号铝合金子母弹经微弧氧化处理后,涂层能够耐2000 ℃的高温气流冲击,20 s 不发生脱落[10]。采用硅酸盐电解液体系对铝硅合金进行微弧氧化处理后,涂层主要由莫来石、α-Al2O3、γ-Al2O3相组成,55 μm 厚的微弧氧化层使基体耐热温度提高约67 ℃,耐热试验时长提高1 倍[11]。尽管微弧氧化层表面一般为多孔结构,但内层相对致密,能够有效抑制高温下氧向基体的扩散,提高基体的抗高温氧化性。在γ-TiAl 合金表面制备的微弧氧化层在800 ℃温度下,100 h 后的氧化增重仅为基体的8.9%,氧化激活能从基体试样的247.79 kJ/mol增加到574.41 kJ/mol[12]。此外,通过调节电解液组分和添加无机颗粒,可进一步提升涂层的热防护功能。例如向微弧氧化电解液中添加纳米ZrO2颗粒,可进一步提升微弧氧化层的隔热能力,这可能与陶瓷层中存在的大量四方相m-ZrO2阻碍热量的传递有关[13]。由此可见,采用微弧氧化技术构筑的陶瓷涂层具有高结合强度、低热导率和良好的抗高温氧化性的特点,以上这些特性显示出微弧氧化技术作为镁合金抗燃热防护涂层的应用潜力[14]。
在探索微弧氧化层抗点燃热防护功能的同时,由于电解液成分和浓度对微弧氧化层性能的贡献至关重要,因此文中以AZ91D 镁合金为基体,选取硅酸盐电解液体系,考察添加不同浓度的K2ZrF6对微弧氧化膜微观结构和相组成的影响,并探究膜层成分和结构变化对微弧氧化层抗点燃性能的作用规律。
1 实验
1.1 材料及涂层制备
实验材料为铸态AZ91D 镁合金,其化学成分(质量分数)为:Al 8.5%~9.5%,Zn 0.45%~1.0%,Mn 0.17%~0.27%,Si≤0.05%,Cu≤0.025%,Ni≤0.001%,Fe≤0.004%,其余为 Mg。试样尺寸为 25 mm×25 mm×10 mm。微弧氧化处理前,用400#砂纸对试样进行打磨,去除表面的氧化层,随后用酒精进行超声清洗,以除去表面污染物,干燥后,用铝导线将其悬挂浸入电解槽中,作为阳极。不锈钢电解槽作为阴极。
采用GGMF 型脉冲电源对镁合金样品进行微弧氧化处理,微弧氧化电解液成分见表1。工作过程中,通过冷却系统控制电解槽温度在35 ℃以下。微弧氧化的电流密度为4 A/dm2,频率为500 Hz,占空比为30%,氧化时间为10 min。涂层制备完成后,进行超声清洗,干燥。

表1 不同K2ZrF6 浓度的微弧氧化电解液Tab.1 The micro-arc oxidation electrolytes with different content of K2ZrF6
1.2 结构表征
采用FEI Quanta 250 扫描电子显微镜对涂层的表面及截面形貌进行观测,并采用Image J 软件对背散射模式下涂层的表面孔隙率和平均孔径进行统计。使用LANDTEK 型粗糙度仪测量涂层的表面粗糙度,取样长度为2.5 mm,选择正反面5 个不同区域进行测量,取其平均值为涂层的粗糙度。采用 DMAX-RB型X-ray 衍射仪对涂层物相进行分析,采用Cu Kα射线,扫描角度为20°~80°,扫描步长为0.02°。
1.3 抗点燃性能测试
为衡量镁合金被火焰引燃的难易程度,对其进行点燃性能测试,并以被点燃时间作为评价指标。测试方法参考美国联邦航空管理局FAA(Federal Aviation Administration)阻燃性能测试方法[15],采用自行设计的实验设备,如图1 所示。该测试装置包括一个提供热源的火焰燃烧器和一个固定测试样品的夹具。25 mm×25 mm 试样面作为火焰加热面,试样完全嵌入由隔热材料制成的夹具中,减少试样与夹具间热量的传递。燃烧器产生的火焰截面近似呈直径为25 mm的圆形,能够完全覆盖样品的加热面。火焰燃烧器口与试样表面的距离为70 mm,实验前用热电偶测量该受火点的温度,约为(1090±20) ℃。在试样背面中心处钻有深8 mm 的ϕ0.8 mm 小孔,用于插入测温的热电偶。实验过程:将测试试样安装在固定夹具上,并插好热电偶,打开火焰加热器,待火焰稳定(30 s 左右)后,将夹具移动到设定位置,加热试样,待试样被点燃后,立即关闭火焰燃烧器,记录下试样的起燃时间和温度。

图1 镁合金抗点燃性能测试装置Fig.1 The ignition resistance torch test apparatus of magnesium alloys
2 结果及分析
2.1 K2ZrF6对镁合金微弧氧化过程的影响
在恒定电流工作模式下,采用不同K2ZrF6浓度的电解液在微弧氧化处理时的电压-时间曲线如图2所示。电压-时间曲线的总体变化趋势没有因K2ZrF6的加入而发生明显的变化,主要分为3 个阶段。在第一阶段(0~40 s),各电解液的电压均呈线性增加的趋势,且起弧电压随着K2ZrF6浓度的升高而逐渐降低。其中,不添加K2ZrF6时的起弧电压约为250 V,而添加20 g/L K2ZrF6时的起弧电压降低到185 V。这是因为向电解液中加入K2ZrF6后,提高了溶液的电导率,而微弧氧化初期产生的微弧等离子体是基体表面的阻抗层失稳击穿所产生的。根据Gouy-Chapman双电层模型[16]可知,在相同工作电压下,高电导率电解液的等效电阻相对减小,使得其两端的分压相对较小,而基体表层击穿介质所分得的电压必然相对较大,分配在膜层上的能量也更高,使得膜层在相对较低的电压下就能被击穿,导致击穿电压下降。在第二阶段(40~360 s),电压增长速度变缓,呈非线性上升的趋势,基体表面的微弧氧化层被击穿,并在高温电弧的作用下不断熔融、冷却、沉积,膜层厚度逐渐增大。在第三阶段(360~600 s),各电解液的电压增长速率明显变得更慢,曲线逐渐趋于水平,最后达到380~415 V 的电压稳定平台。该阶段内的同一时刻,两极间的电压随着电解液中K2ZrF6浓度的增加而稍有增大,添加20 g/L K2ZrF6的电解液的终止电压最高,为415 V,而不添加K2ZrF6的电解液的终止电压最低,为380 V。这可能是由于K2ZrF6在碱性电解液中水解,产生负电性Zr(OH)4颗粒,吸附在试样表面,参与反应,并减小膜层缺陷,使得膜层电阻增大[17]。同时,由于K2ZrF6还会起到增加成膜速率的作用,同样会导致终止电压的升高。这是由于在恒流模式下,随着膜层厚度的不断增大,电流密度需要靠电压的升高来保持稳定[18]。由表2 所列出的膜层厚度可知,在相同处理时间内,K2ZrF6含量更高的电解液所形成的微弧氧化层相对更厚。

表2 膜层的平均尺寸、孔隙率、厚度和表面粗糙度Tab.2 The average size of micro-pores, porosity, thickness and surface roughness of MAO coatings
2.2 微弧氧化膜的微观形貌
使用相同工艺参数在不同K2ZrF6浓度的电解液中所形成的微弧氧化膜的表面形貌如图3 所示。各浓度条件下制备的微弧氧化膜,表面都存在尺寸不一的微孔,这些微孔与微弧氧化放电过程中能量分布不均有关。当微弧氧化进入等离子体放电阶段后,会产生高温高压,使镁合金基体与电解液发生反应,生成熔融物。这些熔融物沿着放电通道不断向外喷射,并在电解液的淬冷作用下凝固在试样表面,形成类火山口状的多孔结构。由表2 的统计结果可知,随着K2ZrF6浓度的增加,微弧氧化膜表面孔径尺寸减小。在添加15 g/L 的K2ZrF6时,孔隙率最低,在添加10 g/L 的K2ZrF6时,表面粗糙度最高,随后呈先降低、后上升的变化趋势。由图2 可知,添加K2ZrF6的电解液的终止电压更高,说明等离子体放电强度也相对更强,从而使每次放电的熔融产物更多。这有利于提高熔融产物对放电通道和表面微孔的填充能力,表面熔融填充痕迹也更为明显(如图3c—e 所示),因此表面孔径和微孔数目都有所降低。另一方面,随着从放电通道喷射出的熔融产物的增多,这些熔融产物会在微孔区域周围冷凝,相互堆积,形成较大凸起,因此使得粗糙度明显增大。此外,K2ZrF6在碱性电解液体系中会与OH–反应,生成呈负电性的Zr(OH)4胶体颗粒[19]。在电场作用下,这些呈负电性的Zr(OH)4胶体颗粒会向阳极表面迁移,吸附在样品表面参与反应,或是沉积在微孔周围,甚至填充部分微孔(如图3d、e 所示),从而提高膜层的致密性,这也会导致终止电压的升高。但当K2ZrF6的质量浓度由15 g/L 提高到20 g/L时,终止电压并没有明显提高,而氧化膜的孔隙率和粗糙度却增大。这可能是因为,此时电解液中处于悬浮状态的负电性Zr(OH)4胶体颗粒已达到饱和,再增加K2ZrF6浓度,所形成的颗粒更多地会沉淀在电解槽底部,而且电解液中的K+、F-等导电离子浓度过高,会使微弧氧化过程变得更加剧烈,反而不利于形成致密的微弧氧化层。

图3 不同K2ZrF6 浓度下制备的微弧氧化膜的表面形貌Fig.3 Surface morphologies of MAO coatings prepared by different K2ZrF6 concentration

图4 不同K2ZrF6 浓度下制备的微弧氧化膜的截面形貌Fig.4 Cross-sectional morphologies of MAO coatings prepared by different K2ZrF6 concentration
不同K2ZrF6浓度下制备的微弧氧化膜与基体间呈锯齿状界面,如图4 所示。这表明膜层是在基体上原位生长的,并与基体呈冶金结合。随着电解液中K2ZrF6浓度的增加,膜层的厚度也随之增大,添加15 g/L 的K2ZrF6制备的微弧氧化层与未添加K2ZrF6的涂层相比,厚度增大了1 倍。此外,由微弧氧化电压-时间曲线可知,微弧氧化层的生长主要发生在第二阶段,且含有高浓度K2ZrF6的电解液起弧电压也更低,膜层在相对较低的电压下就能被击穿,降低了微弧氧化过程的能量消耗,同时放电击穿的次数更多,加速了微弧氧化层的生长。另一方面,随着电解液中K2ZrF6浓度的增加,涂层中的通孔缺陷和裂纹逐渐减少,致密性得到提升。
微弧氧化层在生长过程中存在3 种不同的放电类型[14],即涂层表面微小放电(A-type)、涂层孔洞或裂纹深处-电解液界面放电(B-type)、基体-涂层界面-电解液界面放电(C-type),如图5 所示。微弧氧化层在不同生长阶段的放电类型不同,在达到起弧电压后,微弧氧化进入到第二阶段。在该阶段,涂层表面薄弱位置最先被击穿,形成熔融放电通道,在镁合金基体-氧化层-电解液界面产生剧烈放电,放电类型以C 型为主,并伴有较弱的A 型和B 型等离子体放电。在第三阶段,电压升高速度变缓,膜层较厚,且部分放电通道已被堵塞。该阶段C 类放电频率虽然降低,但强度增加,在表面形成直径较大的孔洞。由图4 可知,在未添加K2ZrF6和添加5 g/L K2ZrF6的微弧氧化膜中,主要存在B 类放电所形成的大孔径孔洞和C 类放电所形成的通孔。随着K2ZrF6浓度的升高,C 类放电所形成的通孔消失,B 类放电形成的大孔径孔洞部分被封闭。K2ZrF6能够提供充足的 F–,与SiO32–、OH–等阴离子相比,F–更容易被Mg 基体所吸附,迁移到膜层内部,在一定程度上起到了封闭微孔,提高膜层致密度的作用[20]。

图5 微弧氧化层表面不同位置放电模式Fig.5 The diagram of discharge modes at different positions on the surface of MAO coatings
2.3 微弧氧化膜的成分及相组成
不同K2ZrF6浓度下制备的微弧氧化膜的物相组成如图6 所示。未添加K2ZrF6的膜层由MgO 和Mg2SiO4相组成,而含有K2ZrF6制备的微弧氧化层XRD 图谱中还出现了MgF2和ZrO2的衍射峰。由此可见,K2ZrF6参与了微弧氧化过程,并且以MgF2和ZrO2相的形式进入膜层当中。
不同膜层表面的EDS 定量结果见表3。不添加K2ZrF6的基础电解液制备的微弧氧化层表面主要以Mg 和O 元素为主,并含有少量源于基体的Al 元素和电解液的Si 元素。结合图4 的XRD 图谱发现,除基体镁的衍射峰外,MgO 衍射峰最强,说明膜层主要由MgO 组成。加入K2ZrF6后,膜层中元素含量有所变化,F 元素含量明显增多,同时Zr 元素含量小幅提升。结合XRD 结果可知,F 元素和Zr 元素分别以MgF2和ZrO2形式存在于膜层中,且MgF2含量较多,而ZrO2含量较少。此外,膜层中原有的O、Mg、Si 等元素含量有所降低,尤其是O 元素含量明显降低,说明膜层中原有的部分MgO 被MgF2所取代。当K2ZrF6添加量超过15 g/L 时,再增加其浓度,膜层表面元素含量变化不明显,Zr 的质量分数由3.78%增加到4.91%。

图6 不同K2ZrF6 浓度下微弧氧化膜的XRD 图谱Fig.6 XRD pattern of the MAO coatings formed in electrolytes with addition K2ZrF6

表3 不同K2ZrF6 浓度下制备的微弧氧化层表面元素含量Tab.3 Element contents of the MAO coatings by different K2ZrF6 concentration wt.%
由图7 可以看出,加入15 g/L 的K2ZrF6后的微弧氧化层中,F 元素的含量高于除基体Mg 外的其他元素,且内层中的含量相对较高。结合微弧氧化层的截面组织(图4c)发现,内层和最外层这两个致密性较高的区域也是F 元素富集的区域。这是因为F–相比电解液中的OH–和SiO32–等阴离子更容易与镁产生化学吸附,从而优先形成内层致密层[21]。同时F原子的半径小于大多数元素,更容易克服已形成的氧化层的阻碍,迁移到膜层内部,因此F 元素在整个膜层中的分布相对较为均匀[22-23]。Mg 和Si 元素含量在膜层内的分布维持在一个相对稳定的水平。O 元素在外层分布较多,主要来自电解液中的OH–和O2,而Mg 对OH–的吸附弱于F–,因此外层O 元素含量多于内层,并以MgO 和ZrO2的形式存在于膜层内。Zr元素主要分布在膜层外层,这是因为在碱性电解液中,K2ZrF6反应生成带负电的Zr(OH)4颗粒,在电场作用下向阳极表面迁移,并吸附在表面,在等离子放电的高温环境下生成ZrO2[24]。
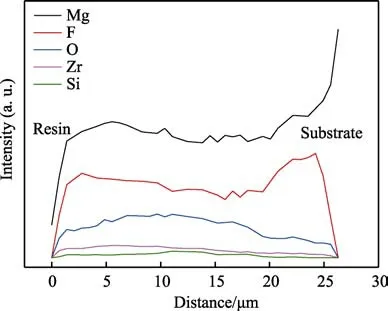
图7 含15 g/L K2ZrF6 制备的微弧氧化膜截面元素分布Fig.7 Element line analysis of MAO coating formed in electrolyte with addition 15 g/L K2ZrF6
2.4 微弧氧化层的抗点燃性能
图8a 给出了AZ91D 镁合金基体和不同K2ZrF6浓度下制备的微弧氧化层试样在火焰加热条件下的起燃时间和温度。结果表明,镁合金基体在火焰加热52 s 后发生燃烧,此时基体温度为549 ℃,处于固液两相区之间,与目前文献所报道的温度接近[25]。而微弧氧化层试样的起燃时间明显增加,且起燃温度均发生在液相区(t液>598 ℃)。图8b 为试样温度在固相线(t固<437 ℃)以下时的温度-时间曲线,可以看到,微弧氧化层试样的温度明显低于无微弧氧化层试样,且15、20 g/L K2ZrF6的试样温度相对较低。主要原因是,微弧氧化层具有较低的热导率,能够阻碍热量向基体的传递,并且膜层中ZrO2相的引入会进一步增强其隔热性能[26]。此时的微弧氧化膜仍保持完整性,起到很好的热防护作用,延缓了高温火焰对基体的破坏。当试样温度升高到液相线温度以上后,微弧氧化层的隔热效果并不明显。主要原因是,此时微弧氧化层遭到不同程度的破坏,导致隔热效果降低。
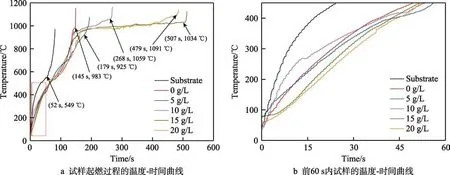
图8 AZ91 镁合金及微弧氧化层的升温曲线Fig.8 The temperature rise curve of AZ91 magnesium alloy and MAO coatings: a) temperature-time curve of sample ignition process; b) temperature-time curve of the sample in the first 60s

图9 不同K2ZrF6 浓度下的微弧氧化膜起燃前的微观组织形貌Fig.9 The morphology of MAO coatings with different K2ZrF6 contents before ignition: a) 0 g/L surface; b) 0 g/L section; c) 5 g/L surface; d) 5 g/L section; e) 10 g/L surface; f) 10 g/L section; g) 15 g/L surface; h) 15 g/L section; i) 20 g/L surface; j) 20 g/L section
此外,对比添加不同含量K2ZrF6制备的微弧氧化层试样的起燃时间可见,未添加K2ZrF6的电解液所获得的MAO 层的起燃时间仅为145 s,抗燃烧性能较差;而引入15~20 g/L 的K2ZrF6后,由于MAO层的致密度提高,使点燃时间明显增加,抗燃烧性能得到提高。镁合金微弧氧化层燃烧前的表面和截面形貌如图9 所示。当微弧氧化层试样的加热时间大约为0.5 倍的点燃时间时(t0≈70 s,t5≈90 s,t10≈135 s,t15≈240 s,t20≈250 s),试样表面均遭到不同程度的破坏。对于0~10 g/L K2ZrF6制备的微弧氧化层,表面形成了大量直径在300 μm 左右的球状颗粒和不规则孔洞(见图9a、c、e)。这是因为当镁合金从固态转变为液态时,会使镁的蒸气压增大几个数量级[27],内部形成的镁蒸气通过微弧氧化层内的通孔结构与外界氧气接触,发生剧烈的氧化反应,同时释放更多热量,导致局部热应力进一步增大,最终导致微弧氧化层破裂,熔化的基体从这些破损的孔洞溢出。溢出的液态镁合金在表面张力的作用下,凝结成球状液滴(见图9e),并在高温火焰作用下,表面破裂,形成起燃点,这些球状颗粒燃烧后脱落,形成烧蚀孔洞(见图9a)。从图9b 能明显看到微弧氧化层中的通孔在火焰冲击作用下完全破碎,镁合金基体沿破碎的通孔向外界扩散,但此时微弧氧化层脱落现象并不明显。图9d、f 与图9b 情况类似,可看到靠近基体的一侧由于镁合金以液态或气态形式溢出后所残留的空隙。此外,对于添加5~10 g/L K2ZrF6所制备的微弧氧化层,涂层表面能观察到以临界烧蚀孔洞/氧化凸起点为中心,向三个方向扩展的裂纹(见图9c、e)。这类裂纹一部分为微弧氧化层原有的微裂纹在火焰冲击所产生的热应力作用下扩展所致,而当这些裂纹扩展至较大孔隙处,裂纹尖端能量足够大时,会在孔隙表面能量薄弱处产生新的裂纹,并继续扩展[28]。因此,微弧氧化层中较大的孔隙会加速这些裂纹的扩展,导致涂层失效。此外,在10 g/L K2ZrF6的电解液中制备的微弧氧化层,表面粗糙度最大,表层起伏程度剧烈,增大了火焰与涂层表面的接触面积,导致基体温度升高得更快,限制了微弧氧化层抗燃烧性的进一步提升。
当K2ZrF6质量浓度增高至15~20 g/L 时,在高温火焰长时间(240~250 s)的冲击下,涂层表面出现撕裂状裂纹。放大观察裂纹表面形貌发现,由于基体已经熔化变形,使得裂纹扩展不共面,呈阶梯状,新形成的不同裂纹面汇合在一起,形成河流状裂纹群(见图9g、i),导致基体暴露在高温火焰下,逐渐形成瘤状颗粒。这些瘤状颗粒会不断堆积长大,最终成为起燃点。通过观察截面形貌(图9h、j)发现,此时破损位置附近的微弧氧化外层的疏松多孔层已部分或者完全脱离基体,而微弧氧化内层的致密层仍然很好地附着在基体表面,提供一定的保护作用。
3 结论
1)在硅酸盐电解液中添加K2ZrF6形成的微弧氧化膜主要由MgO、Mg2SiO4、MgF2和ZrO2组成,K2ZrF6的加入有利于MgF2的生成,且F–更容易迁移到微弧氧化膜的内层。同时,膜层的致密性和厚度得到显著提高,粗糙度随着K2ZrF6含量的增加,呈先升高、后降低的趋势。
2)K2ZrF6浓度对微弧氧化膜抗点燃性的影响主要体现在对其致密性的影响上,内层的致密结构能够一定程度阻碍熔融液态镁和镁蒸气向外的扩散,减缓氧化反应的程度,提高涂层的抗点燃性。
3)添加0~10 g/L K2ZrF6的微弧氧化层的致密性较差,涂层中较大的孔隙在高温火焰作用下,会形成烧蚀孔洞和纵向扩展裂纹;添加15、20 g/L K2ZrF6的微弧氧化层具有更为致密的内层,在火焰冲击过程中,不容易形成烧蚀孔洞,直到基体因完全熔化而变形塌陷,才出现横向扩展裂纹,涂层的抗点燃性更好。
猜你喜欢
金属热处理(2022年8期)2022-11-17
物理学报(2022年2期)2022-02-17
山东冶金(2019年5期)2019-11-16
表面工程与再制造(2019年1期)2019-05-11
科学中国人(2017年35期)2017-06-08
电镀与环保(2016年2期)2017-01-20
当代化工研究(2016年6期)2016-03-20
电源技术(2016年9期)2016-02-27
中国资源综合利用(2016年7期)2016-02-03
中国铸造装备与技术(2015年5期)2015-12-10
