
HSO3−强化Fe3+/S2O82−降解水中双氯芬酸
2021-07-23 01:01王鸿斌刘义青付永胜
中国环境科学 2021年6期
冯 姝,王鸿斌,2,刘义青*,付永胜
HSO3−强化Fe3+/S2O82−降解水中双氯芬酸
冯 姝1,王鸿斌1,2,刘义青1*,付永胜1
(1.西南交通大学地球科学与环境工程学院,四川 成都 611756;2.成都大学建筑与土木工程学院,四川 成都 610106)
采用HSO3−强化Fe3+/S2O82−降解水中双氯芬酸(DCF),考察了溶液初始pH值,Fe3+、HSO3−和S2O82−用量,溶解氧对HSO3−/Fe3+/S2O82−体系降解DCF的影响;通过自由基淬灭实验,识别了体系中主要的活性物种;最后,探讨了DCF在该体系中的降解产物和转化路径.结果表明:HSO3−可以明显促进Fe3+/S2O82−对DCF的降解,初始pH 4.0时,DCF降解效果最佳.DCF的降解速率随Fe3+或S2O82−浓度的增大而增大;适量增加HSO3−浓度可提高DCF的降解,而过量的HSO3−对DCF降解有一定抑制作用.在通入氮气条件下,DCF去除率仅下降10.4%,无明显的抑制作用.自由基抑制实验表明,该体系含有SO4•−、HO•和SO5•−3种活性自由基,其对DCF降解的贡献率分别为83.0%、12.8%和4.3%.在HSO3−/Fe3+/S2O82−降解DCF的反应中共检测出4种产物,据此提出DCF可能的转化路径为:羟基化、脱羧基、脱水和甲酰化反应.
双氯芬酸;亚硫酸氢盐;过硫酸盐;铁离子;硫酸根自由基
双氯芬酸(DCF)作为新兴污染药物,在地下水和地表水中被频繁检测出[1-2].研究表明,痕量的DCF会对水生生物产生不良影响,对生态系统和人类健康构成潜在威胁[3],因此,DCF的水处理技术受到越来越多的关注.近年来,基于过硫酸盐(PS)的高级氧化技术已发展成为解决水中难降解有机污染物的有效方法.PS在室温下具有低反应性,通常可通过加热[4-6]、过渡金属[7-10]、紫外光(UV)[11-13]、碱[14-15]等方式活化,生成高反应性物种,如硫酸根自由基(SO4•−)和羟基自由基(HO•),进一步降解水中有机污染物.通过研究9种不同过渡金属离子对PS的激活能力,结果发现Ag+和Fe2+活化PS降解有机污染物效果最好[7].但是,在Fe2+/PS体系中,氧化生成的Fe3+还原为Fe2+的速率较慢,故在实际应用过程中该体系的持续反应能力不足.近年来,盐酸羟胺[16]、腐殖酸[17]、抗坏血酸[18]等还原剂被引入到Fe2+/PS体系中,加速了Fe3+/ Fe2+的氧化还原,进而提高水中有机污染物的降解效率.Fe3+活化PS的效率非常低,故拟引入亚硫酸盐还原剂.已有研究表明,Fe3+可以和HSO3−结合产生硫酸根自由基和Fe2+[19],产生的Fe2+又可以活化PS产生硫酸根自由基.目前,亚硫酸盐联合Fe3+/PS降解水中有机污染物的研究报道较少.因此,本文系统地研究了HSO3−强化Fe3+/PS对水中DCF的降解,考察HSO3−、PS和Fe3+用量、溶液pH值、溶解氧等对HSO3−/Fe3+/PS降解DCF的影响;识别该体系中主要的活性自由基物种;基于UPLC-Q-TOF-MS对DCF降解产物的鉴定,提出了DCF在该体系中可能的转化途径.
1 材料与方法
1.1 试剂
双氯芬酸(DCF,纯度³98%)购自阿拉丁公司;九水硝酸铁(Fe(NO3)3·9H2O)、亚硫酸氢钠(NaHSO3)、过硫酸钠(Na2S2O8)、硫代硫酸钠(Na2S2O3)、浓硫酸(H2SO4),氢氧化钠(NaOH)均为分析纯级,冰乙酸和叔丁醇为色谱纯,购于成都市科隆化学品有限公司;甲醇为色谱级,购自Fisher Scientific公司;反应液和试剂均用去离子水(18MΩ·cm)进行配置.
1.2 氧化实验
所有实验均在250mL 的玻璃反应器中进行,温度控制在25℃.首先将预定量的试剂(DCF、Na2S2O8、NaHSO3)添加到反应器中,利用0.2mol/L NaOH或H2SO4尽快调节初始pH值,再加入Fe3+启动反应.在反应过程中,通过恒温水浴磁力搅拌器(转子转速为600r/min)确保溶液完全混合.溶解氧影响实验中,在开始反应前,预先向反应液中鼓入氮气或空气吹脱溶液15min.反应启动后在特定的时间间隔(0,20,40,60, 90, 120和180s)提取样品(1mL),并立即用0.2mol/L Na2S2O3(0.05mL)淬灭.所有实验至少重复2次,取平均值作图.
1.3 分析方法
DCF浓度利用高效液相色谱仪(Waters 2695)进行测定,具体参数:固定相为C18柱(4.6mm× 150mm,5μm),流动相为1‰乙酸水溶液与甲醇(= 25/75)混合液,流速为1mL/min,检测波长为276nm,柱温30℃,进样体积为20μL.DCF降解产物采用UPLC-QTOF-MS(Waters)检测.具体参数:色谱柱为BEH C18(2.1mm×50mm,1.7μm);流动相为0.1%甲酸水溶液和乙腈混合液,采用梯度洗脱的方式:0~2min,乙腈由10%提高到30%;2~10min,乙腈提高至100%;10~13min,乙腈降至10%,流速为0.5mL/min;进样体积为1 μL;采用电喷雾电离,在正离子模式下采集数据,扫描范围为50~500Da.TOC浓度使用总有机碳分析仪(Elementar, vario TOC)进行测定.溶液pH值使用PHS-3C型pH计进行测定.DCF在HSO3−/Fe3+/PS体系中的降解符合准一级反应动力学模型,其表观降解速率常数(obs)可由式(1)计算:
ln(C/0) = −obs(1)
式中:obs为表观降解速率常数,s−1;为反应时间, s;0和C分别为初始时间和指定反应时间下DCF的物质的量浓度,μmol/L.
2 结果和讨论
2.1 HSO3−强化Fe3+/PS降解DCF
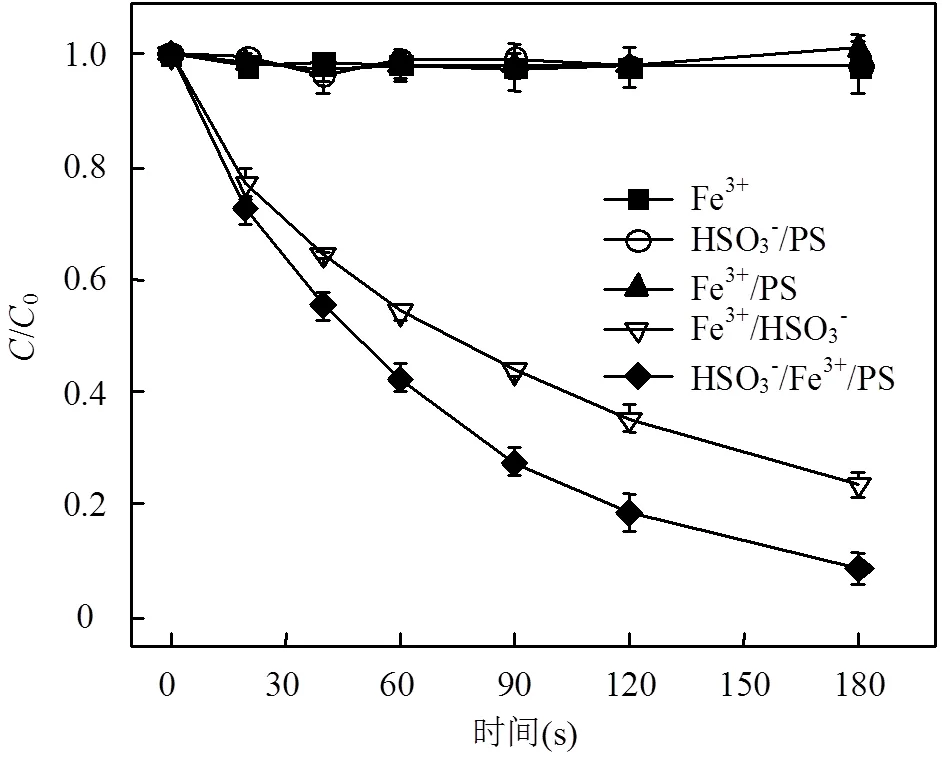
图1 不同体系对DCF降解的影响
[DCF]0= 10μmol/L, [PS]0= 1mmol/L, [HSO3−]0= 200μmol/L, [Fe3+]0= 10μmol/L, pH0= 4.0,= 25oC
如图1所示,在pH = 4.0条件下,单独Fe3+、HSO3−/PS和Fe3+/PS体系均不能有效降解水中的DCF,表明Fe3+不能直接氧化DCF,且Fe3+和低浓度HSO3−对PS活化能力均较差,不能产生活性自由基.然而在Fe3+/PS体系中引入HSO3−,极大地促进了DCF的降解,反应3min 后其去除率达到91.2%,其原因主要为:1)HSO3−是一种还原剂,其加入可将Fe3+还原为Fe2+,作为电子供体的Fe2+可活化PS产生SO4•−,使得DCF快速降解(见式(2)~(4))[20];2)Fe3+可与HSO3−形成FeSO3+配合物,该配体可通过一系列反应使体系中产生SO3•−、SO5•−和SO4•−(见式(2)~ (6)[20]),从而使得DCF降解.由图1可见,在Fe3+/ HSO3−体系中,反应3min后DCF的去除率可达76.5%;3)在HSO3−/Fe3+/PS体系中,Fe3+和Fe2+可以通过一系列氧化还原反应(见式(2)~(7)[20])循环生成,从而促进了SO4•−的生成.
Fe3++ HSO3−→ FeSO3++ H+log= 2.45 (2)
FeSO3+→ Fe2 ++ SO3•−= 0.19s-1(3)
Fe2++ S2O82−→ Fe3++ SO4•−+ SO42−=
2.7´101L/(mol·s) (4)
SO3•−+ O2→ SO5•−= (1.1-2.5)´109L/(mol·s) (5)
SO5•−+ HSO3−→ SO42 −+ SO4•−+ H+= 1.2´
104L/(mol·s) (6)
Fe2++ SO4•−→Fe3++ SO42−= 4.6´109L/(mol·s) (7)
2.2 HSO3−/Fe3+/PS体系中活性自由基的识别
由反应式(8)可知,SO4•−可与水反应产生羟基自由基[21],故在Fe3+/HSO3−/PS体系中,可能同时存在SO4•−、SO3•−、SO5•−和HO•4种活性自由基[22-23].其中SO3•−可快速与氧气反应转化为SO5•−,其对有机物的降解作用可忽略.为鉴定体系中降解DCF的主要活性自由基,并研究甲醇(MeOH)和叔丁醇(TBA)对DCF的抑制作用,引入值(为反应物的物质的量浓度,为反应物与自由基之间的二级反应速率常数)比较反应物之间竞争自由基的能力,见表1.TBA与HO•(= 7.6 × 108L/(mol·s)[24])的反应速率常数远高于SO4•−(= 4.0 × 105L/(mol·s)[25]),可作为HO•的有效抑制剂;而MeOH可同时作为SO4•−(=2.5×107L/(mol·s)[25])和HO•(=9.7×108L/ (mol·s)[24])的抑制剂;另外,MeOH和TBA与SO5•−(<103L/(mol·s)[25])的反应均较慢,故它们不会抑制SO5•−.因此,在一定程度上,TBA和MeOH的抑制实验可验证体系中SO4•−、SO5•−和HO•的存在.
SO4•−+ H2O →HO•+ SO42−+ H+=1.1´101L/(mol·s) (8)
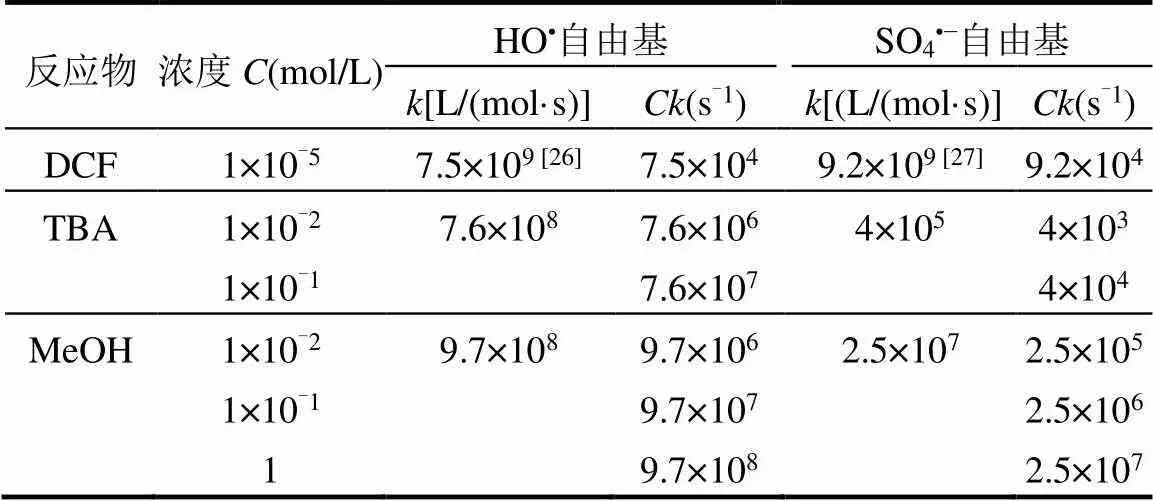
表1 反应物和自由基的Ck值
由图2可看出,与未添加抑制剂相比,加入10mmol/L TBA后DCF降解率降低 14.9%,只能抑制HO•,但不能淬灭SO4•−;该抑制部分可能与HO•的作用有关,因为10mmol/LTBA和HO•的值远大于DCF和HO•的值,而和SO4•−的值则小于DCF.随着TBA浓度增加,其与SO4•−的值接近DCF与SO4•−的值,故它会与DCF竞争体系中的SO4•−,进而进一步抑制DCF的降解.10mmol/L MeOH与SO4•−的值大于100mmol/L TBA与SO4•−的值,故它对DCF去除的抑制作用要强于100mmol/L TBA,如图2所示.随着 MeOH浓度增加至 1mol/L,其与SO4•−的值越来越高,其对DCF降解的抑制作用达到最大.此时,1mol/L MeOH和SO4•−或HO•的值均远大于DCF和SO4•−或HO•的值,但仍有4.1%的DCF被降解.这一结果表明体系中可能存在其他反应物种(即SO5•−),因为 1mol/L MeOH能够完全淬灭SO4•−和HO•.
在加入10mmol/LTBA和1mol/LMeOH的条件下,DCF的表观反应速率常数分别从 4.7×10−3s−1下降到4.1×10−3和2×10−4s−1.据此计算得知, SO4•−、HO•和SO5•−对DCF降解的贡献率分别为83.0%、12.8%和4.3%[28],表明在HSO3−/Fe3+/PS体系降解DCF过程中同时存在SO4•−、SO5•−和HO•3种活性自由基,其中SO4•−为主要活性自由基.
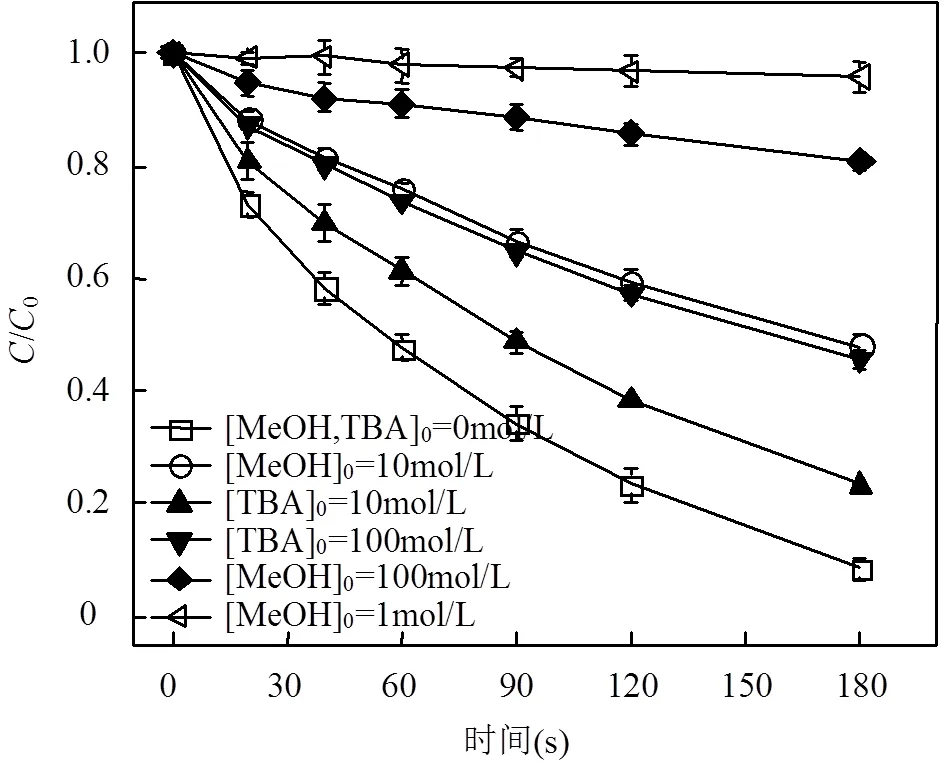
图2 不同自由基淬灭剂对DCF降解的影响
[DCF]0= 10μmol/L, [PS]0= 1mmol/L, [HSO3−]0= 200μmol/L, [Fe3+]0= 10μmol/L, pH0= 4.0,= 25oC
2.3 HSO3−/Fe3+/PS体系降解DCF的影响因素
2.3.1 初始pH值对DCF降解的影响 溶液pH值对HSO3−和Fe3+的形态分布尤为重要,是影响HSO3−/ Fe3+/PS体系的关键性因素,例如:HSO3−在pH>7.0时转化为SO32−的形式存在,而pH 4.0~6.0条件下主要以HSO3−的形式存在,pH<2.0时转为SO2•H2O的形式存在.如图3(a)所示,在pH值大于5.0条件下,DCF几乎不被降解,此条件下Fe3+主要以Fe(OH)2+和Fe(OH)3(aq)形式存在,这2种形态的Fe3+既不能活化PS也不能活化HSO3−[29],以致体系内无活性自由基产生.pH值为 4.0时,体系中存在大量的HSO3−,其与Fe3+通过式(2)形成配体物FeSO3+,该配体物经分解后产生Fe2+和SO3•−(式(3));进一步生成SO5•−和SO4•−,使DCF快速被降解.然而,DCF的降解随着pH值的再度降低而受到抑制,pH值为2.0时,反应3min,仅24.5%的DCF被降解.这主要是由于在该pH值下,HSO3−转化为SO2•H2O,降低了FeSO3+的形成和Fe2+的再生[30-31],进而抑制了SO4•−和SO5•−的生成.
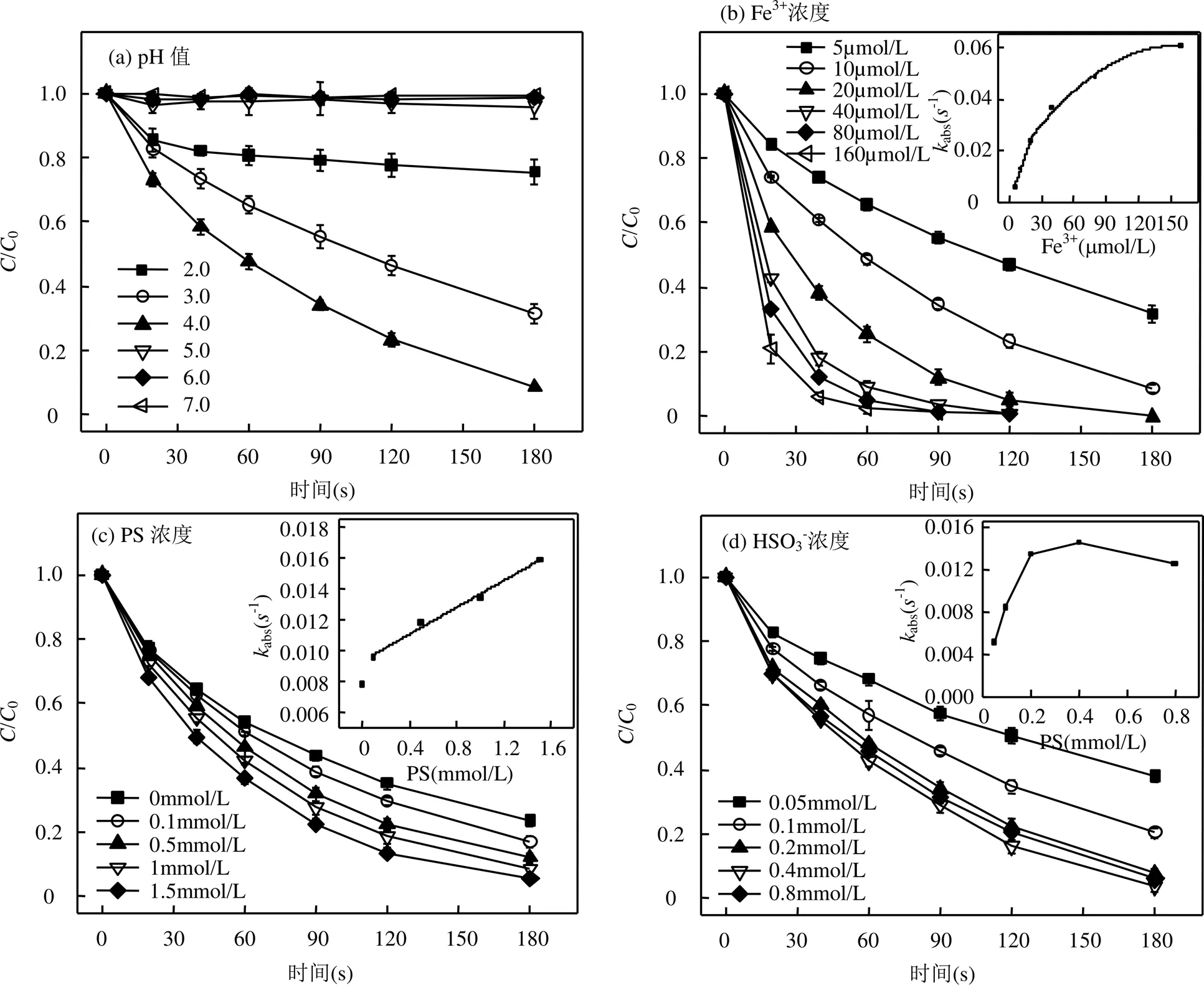
[DCF]0= 10μmol/L, [PS]0= 1mmol/L, [HSO3−]0= 200μmol/L, [Fe3+]0= 10μmol/L, pH0= 4.0,= 25oC
2.3.2 Fe3+浓度对DCF降解的影响 如图3(b)所示,当Fe3+浓度由5μmol/L增加至20μmol/L时,反应3min,DCF的去除率由68.2%提高至100%,继续增加Fe3+浓度至160μmol/L,DCF在90s内完全降解.当Fe3+用量小于20μmol/L时,反应速率常数obs随着Fe3+浓度提高呈线性增大,线性回归斜率为0.0012 (2=0.99).而当Fe3+浓度超过20μmol/L时,虽然obs仍在增加,但它与Fe3+浓度的线性增长关系消失.此现象可解释为,1)Fe3+用量小于20μmol/L时,其浓度的增加加速了对PS和HSO3−的活化作用,产生更多的SO4•−,使DCF降解更快;2)Fe3+浓度在20~ 160μmol/L时,obs增加速率变慢归因于体系中产生过量的Fe2+对SO4•−的淬灭作用(式(7))以及SO4•−的再结合作用(式(9)).王鸿斌等[32-33]在研究Fe2+/PS和Fe2+/HSO3−体系降解DCF的过程中也都观察到了类似的现象.
SO4•−+ SO4•−→S2O82−= (4~8.1)´108L/(mol·s) (9)
2.3.3 PS浓度对DCF降解的影响 如图3(c)显示,当体系中的PS浓度由100μmol/L增加到1.5mmol/L时,DCF的去除率由82.8%增大至94.4%;同时其k和PS浓度有着较好的线性关系,线性回归斜率为0.0093(2= 0.99).这主要是因为PS在Fe2+的活化作用下分解产生强氧化性的活性物种SO4•−(式(4)),促进了DCF的降解,所以PS浓度会直接影响该体系对DCF的去除.
2.3.4 HSO3−浓度对DCF降解的影响 如图3(d),HSO3−用量从0.05mmol/L增加到0.4mmol/L时,DCF降解效率从62.0%提高到96.4%,继续投加HSO3−用量至0.8mmol/L,DCF的去除率下降至94%.当HSO3−投加量小于0.4mmol/L时,obs随着HSO3−浓度的提高逐渐增加,而当HSO3−浓度超过0.4mmol/L时,obs明显减小.此现象可解释为:1) HSO3−用量小于0.4mmol/L时,HSO3−浓度增加会导致FeSO3+浓度增加,进而产生较多的SO4•−和Fe2+,产生的Fe2+又可以活化S2O82−产生更多的SO4•−,因而导致DCF降解速率增快;2)HSO3−浓度大于0.4mmol/L时,obs下降归因于体系中过量的HSO3−对SO4•−的淬灭作用以及SO4•−的再结合作用[34-35].
2.3.5 溶解氧对DCF降解的影响 溶解氧可直接与亚硫酸根自由基反应,从而影响硫氧根自由基之间的链式反应,见式(5)和式(6).因此,考察溶解氧对HSO3−/Fe3+/PS体系降解DCF的影响.如图3(e)所示,与自然搅拌条件相比,曝入空气会促进DCF的降解,使其在2min内即可完全降解.这是因为曝入空气时,反应过程中溶液中的溶解氧浓度要高于自然搅拌条件,反应结束后其残留的溶解氧浓度分别为8.9,7.9mg/L,故曝气加快了SO3•−向SO4•−的转化历程,增加了体系中SO4•−的浓度.一般来说,在缺氧条件下,通过氮气吹扫,溶液中的溶解氧少于2mg/L,会直接抑制SO3•−的氧化,进一步影响硫氧根自由基之间链式反应,减少SO4•−的生成[20].然而在HSO3−/ Fe3+/PS体系中通入氮气对DCF的降解并没有较强的抑制作用,其降解率仅下降了10.4%,这可能是由于:1)缺氧条件下,HSO3−主要以还原作用为主,使体系中Fe3+不断转化为Fe2+,此外,Fe3+与HSO3−形成的配体物亦可分解为Fe2+和SO3•−,继而Fe2+活化PS不断产生SO4•−和Fe3+,故DCF仍有较好的去除率;2)生成的SO3•−与苯胺类有机物的二级反应速率常数约为106L/(mol·s)[25],DCF含有苯胺官能团,故其可能与SO3•−发生反应.
2.4 DCF降解产物及转化途径的鉴定
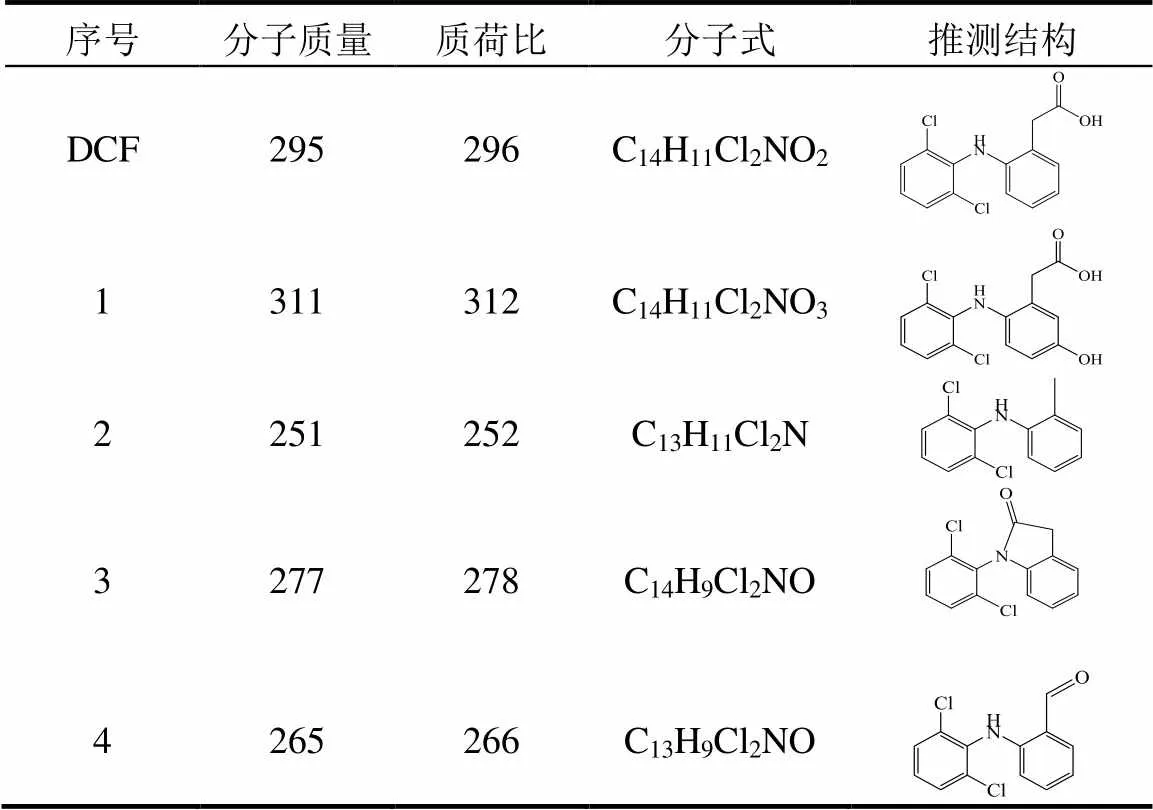
表2 HSO3−/Fe3+/PS体系中DCF的降解产物
在HSO3−/Fe3+/PS降解DCF的过程中共检测到4种反应产物,其分子质量、质荷比()、分子式及可能的结构见表2.基于这些产物提出HSO3−/ Fe3+/PS降解DCF可能的反应机理,主要包括4种不同的降解途径,分别为(1)羟基化反应,(2)脱羧反应,(3)脱水反应和(4)甲酰化反应,具体如图4所示.DCF通过途径(1)、(2)、(3)可以分别生成产物312、252和278.生成的产物252通过途径(4)可以进一步生成产物266.
羟基化反应(途径(1))可能是通过DCF结构中含乙酸基苯环上短暂添加和消除硫酸根离子而发生,从而生成自由基阳离子,该自由基阳离子可通过亲核反应形成羟基化产物312[36].脱羧作用(途径(2))是指DCF的侧链上损失-COOH基团,从而导致252的产生.在脱羧反应之后发生甲酰化反应(途径(4)),产物252中的-CH3基团被氧化为-CHO,从而形成266[37].脱水反应(途径(3))是由DCF上的N原子和-COOH基团偏离而导致羧酸与仲胺分子内酰胺化脱水而引发[38],生成产物278.
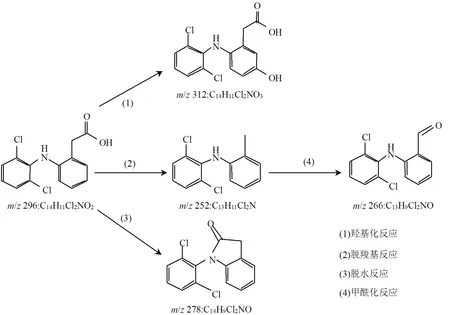
图4 HSO3−/Fe3+/PS体系中DCF可能的降解途径
2.5 HSO3−/Fe3+/PS体系对实际水体中DCF的降解
实际水体包括沱江河河水和西南交通大学犀浦校区犀湖湖水,水质参数如表3所示.由图5(a)可知,在未调节pH值的实际水体中,DCF几乎不被降解,这可能主要是由于pH值的影响.如前文所述,在pH值>5时,DCF几乎不被降解,而河水和湖水的pH值分别为7.6和7.7,在此pH值条件下,Fe3+主要以Fe(OH)3形式存在,此形态的Fe3+既不能活化PS也不能活化HSO3−,以致体系内无活性自由基产生,故DCF不被降解.将实际水体pH值调整为4.0后, HSO3−/Fe3+/PS体系能够降解实际水体中的DCF,但与纯水中的相比,DCF在实际水体中的去除呈现抑制作用,且河水的抑制作用明显强于湖水.由表3可知,河水的碱度、UV254和CODcr均高于湖水,表明河水中的HCO3−和溶解性有机物浓度均高于湖水,而这两种物质能够与体系中的活性自由基反应,从而抑制DCF的降解,故DCF在河水中的去除要弱于湖水,更弱于纯水.此外,本文也研究了DCF在纯水和实际水体中的矿化,结果如图5(b)所示.在反应60min后,纯水中的总有机碳(TOC)衰减了11.6%,表明DCF在HSO3−/Fe3+/PS体系中不能被完全矿化,DCF在自由基的攻击下首先被分解为其他中间产物,这些中间产物的降解矿化可能需要更长的反应时间或者更高的氧化剂用量[36].而河水和湖水中TOC分别降解了17.5%和12.5%,高于纯水,这可能是因为实际水体中本身含有溶解性有机物,这些有机物在自由基的攻击下被氧化降解,从而导致实际水体的矿化率更高.
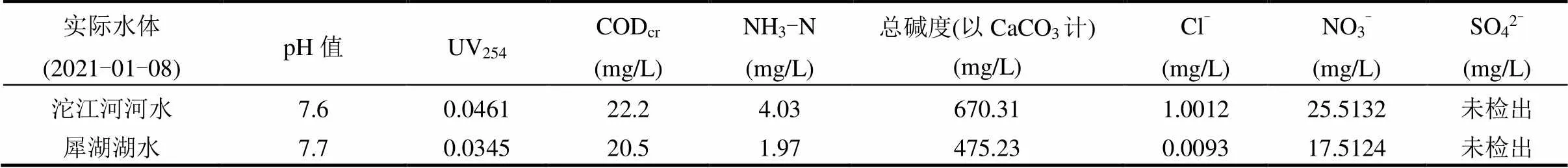
表3 实际水体水质参数

[DCF]0= 10μmol/L, [HSO3−]0= 200μmol/L, [Fe3+]0= 10μmol/L, [PS]0= 1mmol/L,= 25oC
3 结论
3.1 HSO3−可明显强化Fe3+/S2O82−体系对DCF的降解,且pH值为 4.0时,HSO3−/Fe3+/PS对DCF的去除效果最优,但矿化效果不佳,反应60min后TOC仅降解11.6%.
3.2 DCF的降解速率随Fe3+或PS浓度的增大而增大,且与PS浓度成线性正相关;适量增加HSO3−的浓度可提高DCF的降解,而过量的HSO3−对DCF降解有一定抑制作用.在通入氮气条件下,DCF降解率仅下降10.4%,并无明显的抑制作用.
3.3 自由基抑制实验表明,HSO3−/Fe3+/S2O82−体系含有SO4•−、SO5•−和HO•3种活性自由基,其中SO4•−为主要活性自由基.该体系降解DCF的反应中共检出4种产物,由此提出DCF可能的转化路径,分别为羟基化反应、脱羧基反应、脱水反应和甲酰化反应.
3.4 由于实际水体的pH值在中性或者弱碱性,故HSO3−/Fe3+/S2O82−体系对实际水体中的DCF几乎无降解.
[1] Halling S B, Nielsen S N, Lanzky P, et al. Occurrence fate and effects of pharmaceutical substances in the environment-A review [J]. Chemosphere, 1998,36(2):357-393.
[2] Ebele A J, Abdallah M A, Harrad S. Pharmaceuticals and personal care products (PPCPs) in the freshwater aquatic environment [J]. Emerging Contaminants, 2017,3:1-16.
[3] Letzel M, Metzner G, Letzel T. Exposure assessment of the pharmaceutical diclofenac based on long-term measurements of the aquatic input [J]. Environment International, 2009,35(2):363-368.
[4] Waldemer R H, Tratnyek P G, Johnson R L, et al. Oxidation of chlorinated ethenes by heat-activated persulfate: kinetics and products [J]. Environmental Science & Technology, 2007,41(3):1010-1015.
[5] Huang K C, Couttenye R A, Hoag G E. Kinetics of heat-assisted persulfate oxidation of methyl tert-butyl ether (MTBE) [J]. Chemosphere, 2002,49(4):413-420.
[6] 钟 敏,李 孟,卢 芳,等.热活化过硫酸盐联合混凝处理微乳浊液的机理 [J]. 中国环境科学, 2021,41(2):704-712.
Zhong M, Li M, Lu F, et al. Mechanism of thermal-activated persulfate combined with coagulation in the treatment of microemulsion [J]. China Environmental Science, 2021,41(2):704- 712.
[7] Anipsitakis G P, Dionysiou D D. Radical generation by the interaction of transition metals with common oxidants [J]. Environmental Science & Technology, 2004,38(13):3705-3712.
[8] Liang C J, Bruell C J, Marley M C, et al. Persulfate oxidation for in situ remediation of TCE. I. Activated by ferrous ion with and without a persulfate–thiosulfate redox couple [J]. Chemosphere, 2004,55(9): 1213-1223.
[9] Zhang N, Kong X, Zhang M, et al. Study on treatment of methyl- orange in water by derivable oxidation of peroxydisulfate [J]. Journal of Advanced Oxidation Technologies, 2007,10(1):193-195.
[10] 刘美琴,宋秀兰.Fe2+激活过硫酸盐耦合活性炭深度处理焦化废水 [J]. 中国环境科学, 2018,38(4):1377-1384.
Liu M Q, Song X L. Advanced treatment of bio-treated coking wastewater by coupling of ferrous-activated persulfate oxidation and activated carbon adsorption [J]. China Environmental Science, 2018, 38(4):1377-1384.
[11] Mark G, Schuchmann M N, Schuchmann H, et al. The photolysis of potassium peroxodisulphate in aqueous solution in the presence of tert-butanol: a simple actinometer for 254nm radiation [J]. Journal of Photochemistry and Photobiology A: Chemistry, 1990,55(2):157-168.
[12] Gao Y, Gao N, Deng Y, et al. Ultraviolet (UV) light-activated persulfate oxidation of sulfamethazine in water [J]. Chemical Engineering Journal, 2012,195-196:248-253.
[13] 黄丽坤,李 哲,王广智,等.紫外催化过硫酸盐深度处理垃圾焚烧厂渗滤液 [J]. 中国环境科学, 2021,41(1):161-168.
Huang L K, Li Z, Wang G Z, et al. Advanced treatment of landfill leachate by ultraviolet catalytic persulfate [J]. China Environmental Science, 2021,41(1):161-168.
[14] Liang C, Su H W. Identification of sulfate and hydroxyl radicals in thermally activated persulfate [J]. Industrial & Engineering Chemistry Research, 2009,48(11):5558-5562.
[15] Furman O S, Teel A L, Watts R J. Mechanism of base activation of persulfate [J]. Environmental Science & Technology, 2010,44(16): 6423-7428.
[16] 邹 景.羟胺对Fe2+/过硫酸盐体系的强化效能与机理研究 [D]. 哈尔滨:哈尔滨工业大学, 2016.
ZHOU J. Enhanced effectiveness and mechanism of Fe2+/persulfate system with hydroxylamine [D]. Harbin: Harbin Institute of Technology, 2016.
[17] Li D W, Chen D Z, Yao Y Y, et al. Strong enhancement of dye removal through addition of sulfite to persulfate activated by a supported ferric citrate catalyst [J]. Chemical Engineering Journal, 2016,288:806-812.
[18] Curtin M A, Taub I A, Kustin K, et al. Ascorbate-induced oxidation of formate by peroxodisulfate: product yields, kinetics and mechanism [J]. Research on Chemical Intermediates, 2004,30(6):647-661.
[19] Wang S X, Wang G S, Fu Y S, et al. A simple Fe3+/bisulfite system for rapid degradation of sulfamethoxazole [J]. RSC Advances, 2020,10(50):30162-30168.
[20] Liu Z Z, Guo Y Z, Shang R, et al. A triple system of Fe(III)/sulfite/ persulfate: Decolorization and mineralization of reactive Brilliant Red X-3B in aqueous solution at near-neutral pH values [J]. Journal of the Taiwan Institute of Chemical Engineers, 2016,68:162-168.
[21] Herrmann H, A Reese, R Zellner. Time-resolved UV/VIS diode array absorption spectroscopy of SOx•−(= 3, 4, 5) radical anions in aqueous solution [J]. Journal of Molecular Structure, 1995,348:183-186.
[22] Xu J, Ding W, Wu F, et al. Rapid catalytic oxidation of arsenite to arsenate in an iron(III)/sulfite system under visible light [J]. Applied Catalysis B: Environmental, 2016,186(5):56-61.
[23] Chen L, Peng X, Liu J, et al. Decolorization of orange II in aqueous solution by an Fe (II)/sulfite system: replacement of persulfate [J]. Industrial & Engineering Chemistry Research, 2012,51(42):13632-13638.
[24] Buxton G V, Greenstock C L, Helman W P, et al. Critical review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (•OH/O•−) in aqueous solution [J]. Journal of Physical and Chemical Reference Data, 1988,17(2):513-886.
[25] Neta P, Huie R E, Ross A B. Rate constants for reactions of inorganic radicals in aqueous solution [J]. Journal of Physical and Chemical Reference Data, 1988,17(3):1027-1284.
[26] Huber M M, Canonica S, Park G Y, et al. Oxidation of pharmaceuticals during ozonation and advanced oxidation processes [J]. Environmental Science & Technology, 2003,37(5):1016-1024.
[27] Ahmed M M, Barbati S, Doumenq P, et al. Sulfate radical anion oxidation of diclofenac and sulfamethoxazole for water decontamination [J]. Chemical Engineering Journal, 2012,197:440-447.
[28] Liu Y Q, He X X, Fu Y S, et al. Degradation kinetics and mechanism of oxytetracycline by hydroxyl radical-based advanced oxidation processes [J]. Chemical Engineering Journal, 2016,284:1317-1327.
[29] Kolthoff I M, Miller I K. The chemistry of persulfate. I. The kinetics and mechanism of the decomposition of the persulfate ion in aqueous medium1 [J]. Journal of the American Chemical Society, 1951,73(7): 3055-3059.
[30] Graedel T E, Weschler C J. Chemistry within aqueous atmospheric aerosols and raindrops [J]. Reviews of Geophysics, 1981,19(4):505- 539.
[31] Grgićc I, Pozničc M, Bizjak M. S(IV) autoxidation in atmospheric liquid water: The role of Fe(II) and the effect of oxalate [J]. Journal of Atmospheric Chemistry, 1999,33(1):89-102.
[32] 王鸿斌,王 群,刘义青,等.亚铁活化过硫酸盐降解水中双氯芬酸钠 [J]. 环境化学, 2020,39(4):869-875.
Wang H B, Wang Q, Liu Y Q, et al. Degradation of diclofenac by ferrous activated persulfate [J]. Environmental Chemistry, 2020, 39(4):869-875.
[33] Wang H B, Wang S X, Liu Y Q, et al. Degradation of diclofenac by Fe(II)-activated bisulfite: Kinetics, mechanism and transformation products [J]. Chemosphere, 2019,237:124518.
[34] Huie R E, Neta P. Rate constants for some oxidations of S(IV) by radicals in aqueous solutions [J]. Atmospheric Environment, 1987, 21(8):1743-1747.
[35] Huie R E, Clifton C L, Altstein N. A pulse radiolysis and flash photolysis study of the radicals SO2•−, SO3•−, SO4•−and SO5•−[J]. International Journal of Radiation Applications and Instrumentation Part C Radiation Physics and Chemistry, 1989,33(4):361-370.
[36] Liu Y Q, He X X, Fu Y S, et al. Kinetics and mechanism investigation on the destruction of oxytetracycline by UV-254nm activation of persulfate [J]. Journal of Hazardous Materials, 2016,305:229-239.
[37] Pérez-Estrada L A, Malato S, Gernjak W, et al. Photo-Fenton degradation of diclofenac: identification of main intermediates and degradation pathway [J]. Environmental Science & Technology, 2005,39(21):8300-8306.
[38] Zhou T, Feng K, Xiang W, et al. Rapid decomposition of diclofenac in a magnetic field enhanced zero-valent iron/EDTA Fenton-like system [J]. Chemosphere, 2018,193:968-977.
Bisulfite enhanced degradation of diclofenac in Fe3+/persulfate system.
FENG Shu1, WANG Hong-Bin1,2, LIU Yi-Qing1*, FU Yong-Sheng1
(1.Faculty of Geosciences and Environmental Engineering, Southwest Jiaotong University, Chengdu 611756, China;2.School of Architecture and Civil Engineering, Chengdu University, Chengdu 610106, China)., 2021,41(6):2677~2684
The degradation of diclofenac (DCF) by bisulfite enhanced Fe3+/persulfate system was investigated. The influence of pH, Fe3+dosage, HSO3−dosage, persulfate (PS) dosage and dissolved oxygen on DCF degradation in HSO3−/Fe3+/S2O82−system was explored. The main reactive radical species for DCF removal in this system was also identified by scavenging experiments. Finally, The degradation products and transformation mechanism of DCF by HSO3−/Fe3+/S2O82−were evaluated. DCF could be effectively degraded by the introduction of HSO3−in Fe3+/PS process, and the optimal pH was 4.0. The increased initial Fe3+, HSO3−or PS concentration promoted DCF degradation while excessive HSO3−could inhibit its degradation by acting as a SO4•−scavenger. The degradation rate of DCF was only reduced by 10.4% with bubbling nitrogen, and there was no obvious inhibitory effect in this system. According to the radical scavenging experiments, the contribution of SO4•−, HO•and SO5•−to DCF degradation in HSO3−/Fe3+/S2O82−system were calculated to be 83.0%, 12.8% and 4.3%, respectively. Four transformation products were detected using UPLC-Q-TOF-MS. The potential degradation mechanism of DCF was thus proposed showing four reaction pathways including hydroxylation, decarboxylation, dehydration and formylation.
diclofenac;bisulfite;persulfate;ferric ion;sulfate radical
X703
A
1000-6923(2021)06-2677-08
2020-11-04
四川省科技厅重大专项(2018SZDZX0026);中央高校基本科研业务费科技创新项目(2682018CX32)
* 责任作者, 讲师, liuyq@swjtu.edu.cn
冯 姝(1996-),女,四川广元人,西南交通大学硕士研究生,研究方向为水污染控制.
猜你喜欢
材料与冶金学报(2022年2期)2022-08-10
科技视界(2021年21期)2021-08-24
云南化工(2020年11期)2021-01-14
作文成功之路·小学版(2020年6期)2020-07-27
科学与信息化(2020年11期)2020-06-19
当代水产(2020年4期)2020-06-16
当代水产(2020年3期)2020-06-15
科学中国人(2018年8期)2018-07-23
计算机测量与控制(2017年6期)2017-07-01
水利科技与经济(2017年6期)2017-04-28
