
美沙拉嗪肠溶缓释颗粒剂的大鼠药动学及胃肠道分布研究
2021-07-28 07:19张朝绅许子华中国人民解放军北部战区总医院药剂科辽宁沈阳110000
药学实践杂志 2021年4期
胡 北,史 英,张朝绅,许子华 (中国人民解放军北部战区总医院药剂科,辽宁 沈阳 110000)
美沙拉嗪化学名为5-氨基水杨酸,是轻、中度溃疡性结肠炎有效的一线治疗药物。它直接作用于炎症黏膜,可抑制引起炎症的前列腺素合成、抑制炎症介质白三烯的形成而起抗炎作用,具有疗效好及毒副作用小等优点[1-2]。美沙拉嗪口服后在体内被迅速吸收,在肠腔及肝脏中经N-乙酰转移酶代谢为主要产物N-乙酰-5-氨基水杨酸。一般认为,其在肠腔黏膜局部浓度越大越有利于发挥作用[3]。
目前市售的美沙拉嗪药物有美沙拉嗪缓释颗粒剂(Etiasa)、美沙拉嗪肠溶片等剂型,考虑到人们的用药方便和美沙拉嗪的作用部位,课题组开发了美沙拉嗪药物的新剂型,即美沙拉嗪肠溶缓释颗粒剂,以期同时发挥药物肠溶和缓释的作用效果,均匀释放、定位起效,达到更好、更有效的治疗溃疡性结肠炎并减轻毒副作用的效果。为了考察美沙拉嗪新制剂与市售制剂的区别,本实验考察了美沙拉嗪新制剂临床前体内药动学特征,以市售制剂为参比,考察新制剂的相对生物利用度,进行药动学参数的比较评价;并且进一步考察了美沙拉嗪肠溶缓释颗粒剂的制剂学肠溶和缓释特点,评价作用部位释放情况,为该药新制剂提供系统可靠的临床前药动学数据,并为进一步的制剂工艺和处方的优化提供依据。
1 仪器与试药
1.1 仪器
VARIAN 1200L 型液相色谱-质谱联用仪(美国瓦里安公司);Dionex SUMMIT 高效液相色谱仪(美国戴安公司);Jouan CR3i 型多功能高速冷冻离心机(法国Jouan 公司);SAVANT SPD121P SpeedVac型减压离心浓缩仪(北京塞万特公司);XS205 Dual Range 型电子分析天平(梅特勒-托利多公司)。
1.2 试药
美沙拉嗪标准对照品(批号:100546-200902,含量:99.8%)、乙酰美沙拉嗪标准对照品(批号100711-200401,含量:99.3%)、氯唑沙宗标准对照品均由上海市药物(中药)代谢产物研究重点实验室提供;美沙拉嗪肠溶缓释颗粒剂(1 g/袋,批号DV121102-1A)为自研制剂;美沙拉嗪缓释颗粒剂(批号:07444,规格500 mg/袋,Ethypharm 生产,法国)为市售制剂;甲醇(MERK 公司)、乙腈(TEDIA 公司)、甲酸(ROE SCIENTIFIC INC 公司)均为色谱纯试剂;醋酸铵(江苏强盛功能化学股份有限公司)为分析纯;水为去离子水(经Hi-Tech水纯化系统自制)。
1.3 动物
健康、成年SD 大鼠,体重180~220 g,雌、雄数量均等,年龄6~8 周,购自上海斯莱克实验动物有限责任公司[许可证号SCXK(沪)2012-0005]。
2 美沙拉嗪肠溶缓释颗粒剂的药动学研究
2.1 SD 大鼠血浆中目标药物浓度分析方法
2.1.1 色谱条件及质谱检测条件
流动相为甲醇-5 mmol/L 醋酸铵水溶液(85∶15);流速为0.4 ml/min,进样量10 μl[4-5]。
电喷雾离子源,负离子模式检测,多反应监测方式进行一/二级质谱分析。质谱检测工作参数如下:美沙拉嗪、乙酰美沙拉嗪和内标氯唑沙宗Q1(Mass)分别是151.7、193.8、167.8;Q3(Mass)分别是107.9、149.7、131.7;扫描时间0.33s,SIM 宽0.7,Q1 峰宽3.0;Q3 峰宽2.0;Needle-4500V;Shield-25 V;雾化气为空气,48psi;干燥气为空气,300 ℃,20psi;毛细管电压为−35 V;碰撞能分别是13、14、19 V,碰撞气:1.84mTorr;检测器电压为1 650 V[6-7]。
2.1.2 溶液的配制
精密称取美沙拉嗪、乙酰美沙拉嗪和内标氯唑沙宗适量,加甲醇溶解,配制成浓度为5 mg/ml 的标准储备液,然后采用相应的流动相逐级稀释,得到系列混合工作溶液和内标溶液。同法制备质控标准样品的系列混合工作溶液和内标溶液。
2.1.3 血浆样品的处理
取100 μl 血浆样品,加入氯唑沙宗溶液10 μl,涡旋;然后加入甲醇300 μl,涡旋3 min,离心10 min(8 000 r/min);吸取上清液250 μl,再离心5 min(12 000 r/min);吸取上清液100 μl 待测。
2.2 分析方法的确证
2.2.1 特异性
在上述分析方法下,考察样品、内标和内源性物质的干扰情况。结果表明生物样品中的内源性物质不干扰美沙拉嗪和乙酰美沙拉嗪的测定,结果见图1。
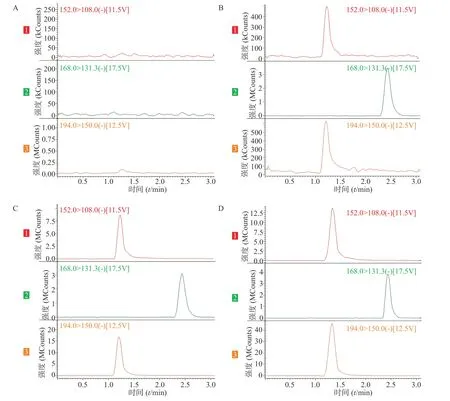
图1 SD 大鼠血浆中美沙拉嗪、乙酰美沙拉嗪的LC-MS/MS 典型谱图
2.2.2 线性范围和最低定量限
精密吸取美沙拉嗪和乙酰美沙拉嗪系列混合标准工作溶液,用空白血浆进行提取,配制成美沙拉嗪的浓度为0.100 2、0.200 4、0.501、2.004、5.01、10.02、20.04 μg/ml 和乙酰美沙拉嗪的浓度为0.020 12、0.050 3、0.201 2、1.006、5.03、10.06、20.12 μg/ml,进样分析后求得美沙拉嗪和乙酰美沙拉嗪的标准曲线方程分别为Y=0.691 4C+0.031 0(r=0.999 3)、Y=3.102 2C+0.022 7(r=0.999 9),线性范围为0.100 2~20.04 μg/ml 和0.020 12~20.12 μg/ml,最低定量限分别为0.100 2 和0.020 12 μg/ml(S/N>5)。
2.2.3 准确度和精密度
按照上述方法配制最低定量限、低、中、高4 个浓度的质控标准样品,美沙拉嗪和乙酰美沙拉嗪质控标准样品浓度分别为0.100 4、0.020 04,0.200 8、0.050 1,2.008、1.002 和16.064、16.032 μg/ml。结果表明,准确度和精密度满足生物基质样品测定要求。
2.2.4 基质效应和绝对回收率
在低、中、高3 个浓度下,美沙拉嗪和乙酰美沙拉嗪在SD 大鼠血浆中基质效应在70%~98%之间,绝对回收率在70%~90%之间,表明内源性物质对被测物质存在一定的抑制或增强的作用,但不影响样品的分析测定。
2.2.5 稳定性
血浆样品在室温下放置4h、−80 ℃保存60 d,处理后在自动进样器内放置24 h,以及3 次冻融循环后样品均保持稳定,RSD 在15%以内。结果表明,各生物基质样品在所考察的时间和条件下稳定,对测定结果的影响可以忽略。
2.3 药动学研究
12 只成年、健康SD 大鼠,雌性、雄性各半,随机分成2 组,每组6 只;分别灌胃给予自研制剂和市售制剂各0.1 g/kg。
分别于给药前及给药后的1、2、3、4、5、6、8、12、24、36 和48 h,从SD 大鼠眼眶取血,置肝素化EP 管中,离心,取上层血浆,置于−80 ℃冰箱保存待测。按照上述样品前处理的方法对生物基质样品进行预处理,并进样分析,计算目标药物浓度[8]。
数据处理:药动学数据以三因素方差分析和双单侧t检验进行统计分析。应用中国药科大学开发的药动学处理软件BAPP2.0 程序计算有关药动学参数。
2.3.1 美沙拉嗪在SD 大鼠体内的药动学参数
采用非房室模型法估算的以美沙拉嗪表征的两组成年、健康SD 大鼠给予自研制剂和市售制剂0.1 g/kg 后的体内药动学参数见表1,药时曲线见图2。
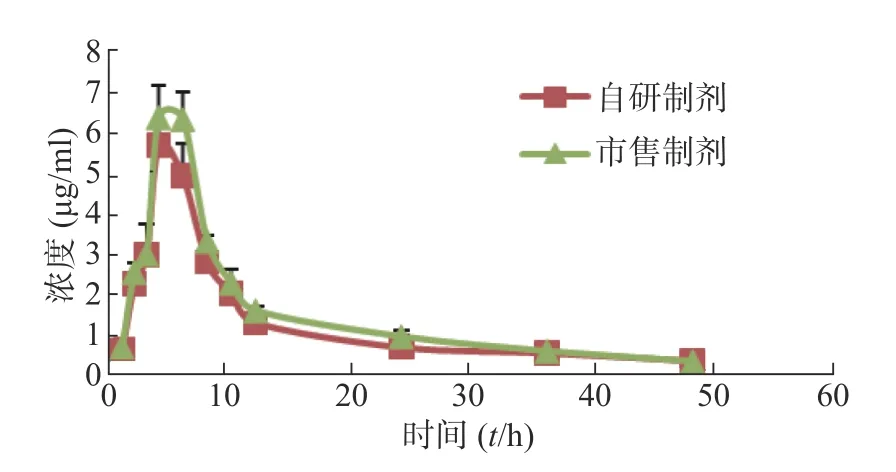
图2 自制和市售美沙拉嗪的药时曲线图
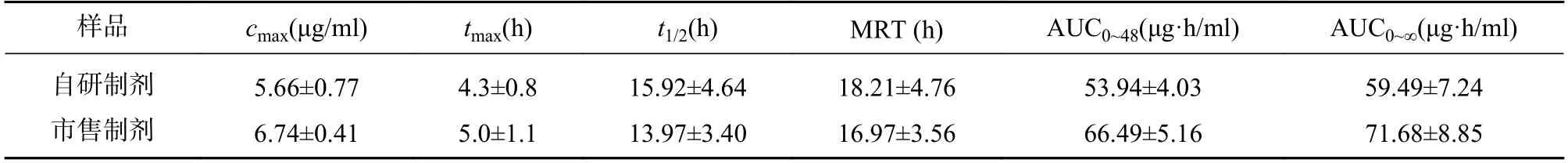
表1 口服给药后体内以美沙拉嗪表征的药动学参数
用梯形面积法(AUC)估算的以美沙拉嗪表征的自研制剂和市售制剂的相对生物利用度为(90.62±9.36)%,即平均生物利用度基本一致。
2.3.2 乙酰美沙拉嗪在SD 大鼠体内的药动学参数
采用非房室模型法估算的以乙酰美沙拉嗪表征的两组成年、健康SD 大鼠给予自研制剂和市售制剂0.1 g/kg 后的体内药动学参数见表2,药时曲线见图3。
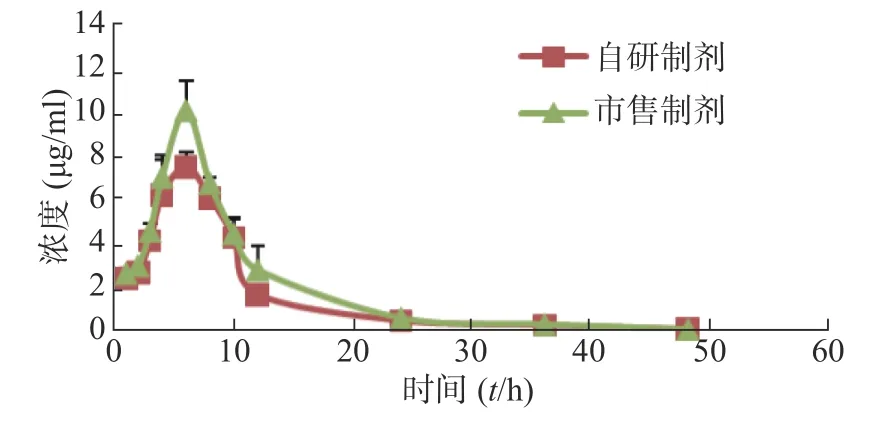
图3 自制和市售乙酰美沙拉嗪的药时曲线图
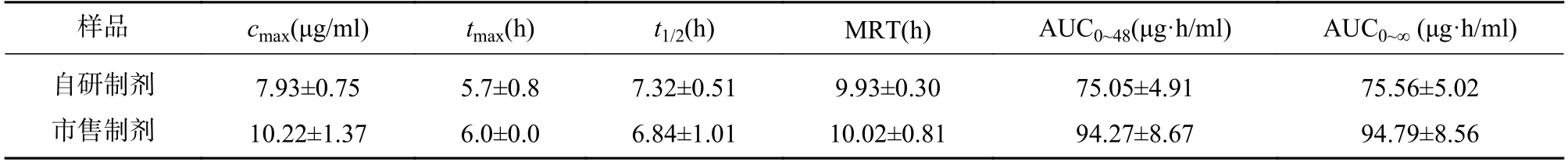
表2 口服给药后体内以乙酰美沙拉嗪表征的药动学参数
采用梯形面积法(AUC)估算的以乙酰美沙拉嗪表征的自研制剂和市售制剂的相对生物利用度为(87.36±10.43)%,即平均生物利用度基本一致。
3 美沙拉嗪肠溶缓释颗粒剂胃肠道分布和药物残留研究
3.1 SD 大鼠胃肠道内容物中目标药物浓度分析方法
3.1.1 胃肠道内容物分析方法
流动相为乙腈-20 mmol/L 醋酸铵水溶液(2∶98);流速1.0 ml/min;检测波长为304 nm,柱温25 ℃。
3.1.2 溶液的配制
精密称取美沙拉嗪适量,加甲醇溶解,配成浓度为5 mg/ml 的标准储备液,采用相应的流动相来逐级稀释,得到系列混合工作溶液和内标溶液。同法制备质控标准样品的系列混合工作溶液和内标溶液。
3.1.3 胃肠道内容物的处理
将SD 大鼠胃肠消化道分段内容物样品在室温下解冻,将药物颗粒数量大于5 粒的内容物样品转移至100 ml 烧杯中,搅拌均匀,再准确加入50 ml去离子水清洗相应的消化道分段,并溶解其内容物;药物颗粒数量小于5 粒的内容物样品转移至10 ml EP 离心管中,搅拌均匀,再准确加入5 ml 去离子水清洗相应的消化道分段,并溶解其内容物[9-10];分别超声溶解各内容物样品20 min,取1 ml 溶液12 000 r/min 离心10 min,分离上清液,再12 000 r/min离心10 min 后,分离上清液待测。
3.2 分析方法的确证
3.2.1 特异性
在上述分析方法下,考察了样品和内源性物质的干扰。结果表明生物样品中的内源性物质不干扰美沙拉嗪的测定,结果见图4~图8。
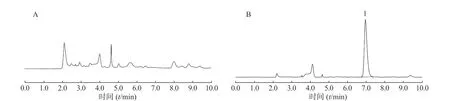
图4 大鼠胃内容物样品中美沙拉嗪的HPLC 图

图5 大鼠十二指肠内容物样品中美沙拉嗪的HPLC 图
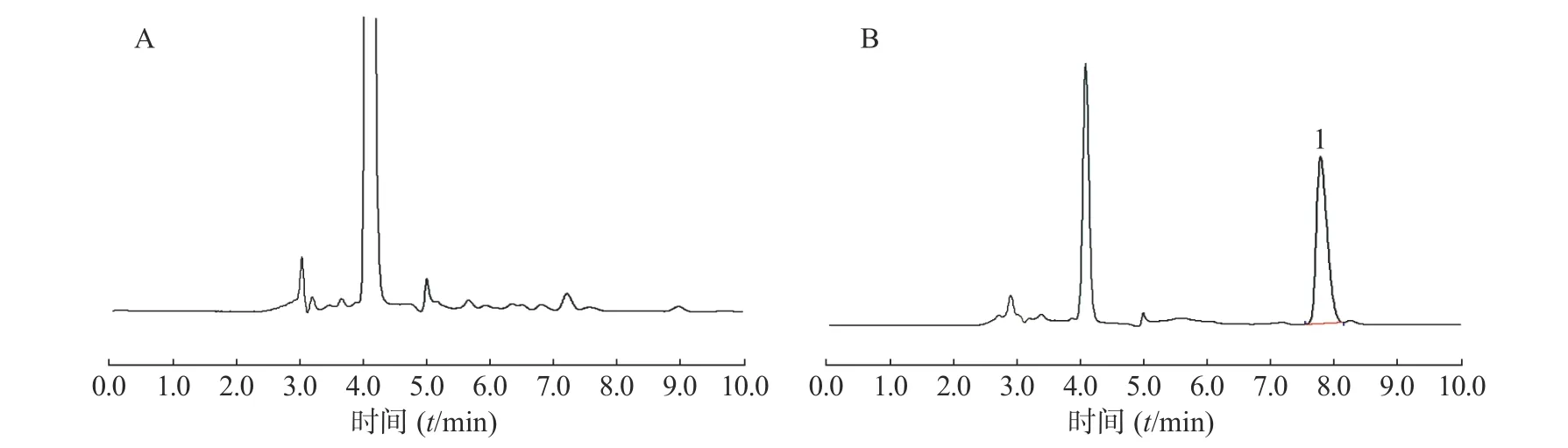
图6 大鼠空肠内容物样品中美沙拉嗪的高效液相色谱图
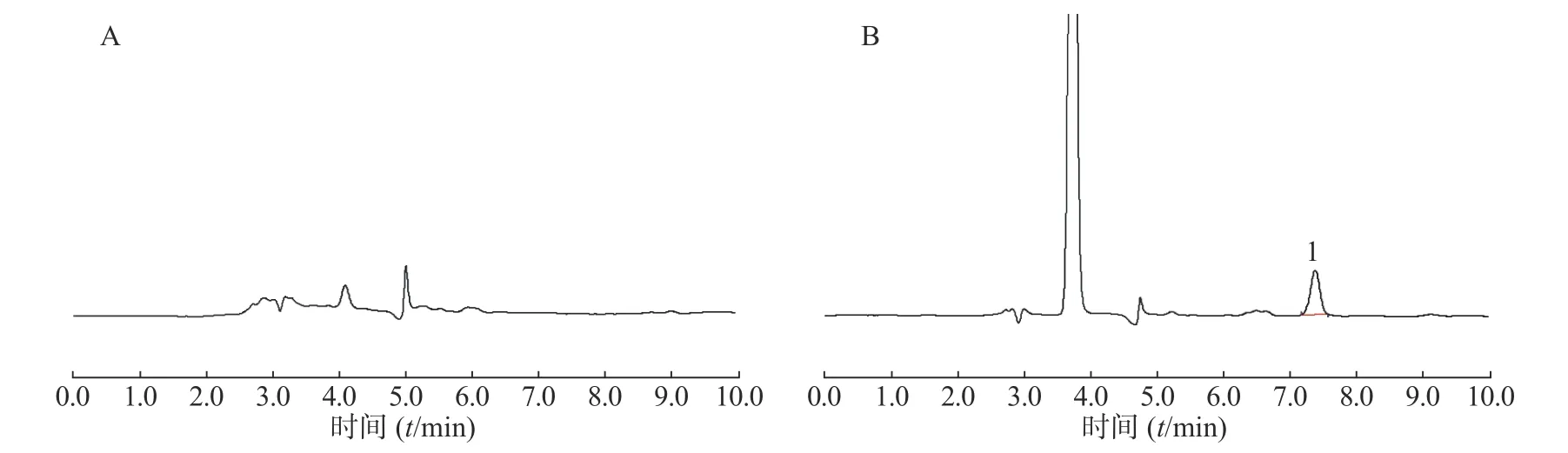
图7 大鼠回肠内容物样品中美沙拉嗪的HPLC 图
图8 大鼠结肠内容物样品中美沙拉嗪的HPLC 图
3.2.2 线性范围和最低定量限
精密吸取适量美沙拉嗪,用空白胃内容物提取上清液,配制成美沙拉嗪浓度分别为0.504 5、1.009、5.045、10.09、20.18、50.45 和100.9 μg/ml 的溶液,进样分析,求得标准曲线方程为Y=0.130 8C+0.005 3(r=0.999 6),线性范围为0.504 5~100.9 μg/ml,最低定量限为0.504 5 μg/ml(S/N>5);用空白肠道内容物提取上清液配制成美沙拉嗪浓度分别为0.513 5、1.027、5.135、10.27、20.54、51.35 和102.7 μg/ml,进样分析后求得标准曲线方程为Y=0.196 1C-0.008 1,r=0.999 9,线性范围为0.513 5~102.7 μg/ml,最低定量限为0.513 5 μg/ml(S/N>5)。
3.2.3 准确度和精密度
按照上述方法配制最低定量限、低、中、高4 个浓度的质控标准样品,SD 大鼠胃内容物美沙拉嗪质控标准样品浓度为0.502、1.004、10.04 和80.32 μg/ml,SD 大鼠肠内容物美沙拉嗪质控标准样品浓度为0.504、1.008、10.08、80.64 μg/ml。结果表明,准确度和精密度满足生物基质样品测定要求。
3.2.4 基质效应和绝对回收率
在最低定量限、低、中、高4 个浓度下,SD 大鼠胃肠道内容物中美沙拉嗪的绝对回收率在95%~99%之间,表明内源性物质对被测物质存在一定的抑制或增强的作用,但不影响样品的分析测定。
3.2.5 稀释效应
本实验生物基质中药物浓度大于标准曲线上限,须经稀释后再测定,故考察了稀释效应。配制高浓度的质控样品,稀释相应倍数至高浓度,按照生物样品前处理方法进样测定,代入标准曲线求得美沙拉嗪浓度,稀释效应采用测定值与理论值RE 值表示。结果表明,样品经稀释后分析结果准确,能满足生物基质样品分析要求。
3.2.6 稳定性
胃肠道样品在室温下放置4 h、−80 ℃长期保存60 d、处理后在自动进样器内放置36 h,以及3 次冻融循环后样品均保持稳定,RSD 在15%以内。结果表明,各生物基质样品在所考察的时间和条件下稳定,对测定结果的影响可以忽略。
3.3 美沙拉嗪肠溶缓释颗粒剂胃肠道分布和药物残留研究
选用60 只成年、健康SD 大鼠,雌雄各半,实验SD 大鼠禁食过夜(禁食15 h)后,随机分成2 大组,分别以100 mg/kg 给予自研制剂和市售制剂;每一大组又随机分成5 小组,每小组6 只,选取给药后2、4、6、8 和12 h 的大鼠胃肠消化道内分布的美沙拉嗪颗粒及药物残留量。另取6 只SD 大鼠(雌雄各半)作为空白对照,同样禁食过夜(禁食15 h)。分别于给药后2、4、6、8 和12 h(每一时间点各6 只SD 大鼠),将SD 大鼠乙醚麻醉,完整取出胃、十二指肠段、空肠段、回肠段和结肠段组织,分段解剖分离药物颗粒及内容物,置于−80 ℃冰箱保存待测。按胃肠消化道分段内容物样品预处理方法操作,测定胃肠消化道内容物中美沙拉嗪的浓度[11-12]。
由胃肠消化道内容物中美沙拉嗪浓度计算而得的自制和市售两种美沙拉嗪缓释颗粒相应的胃肠道美沙拉嗪残留量结果见图9 和图10。

图9 自研制剂在消化道各部位不同时间点的药物残留量
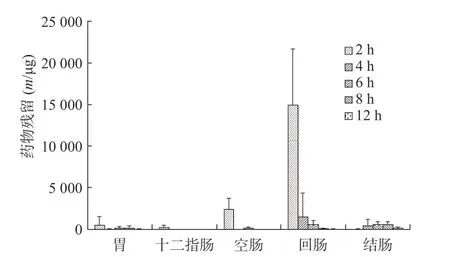
图10 市售制剂在消化道各部位不同时间点的药物残留量
4 讨论
本实验通过大鼠口服给药考察了美沙拉嗪肠溶缓释颗粒剂的药动学特征、相对生物利用度,并与市售制剂的药动学参数进行了比较研究,结果表明,自研制剂口服后给药后吸收和消除过程基本呈线性动力学特征。通过相对生物利用度和药动学参数的统计分析比较,提示自研制剂与市售制剂无显著性差异。
从药物颗粒分布结果看,自研制剂的微丸在SD 大鼠胃肠消化道内具有一定流动性,且包衣对胃肠消化液的耐受性较好,随着SD 大鼠胃肠消化道的蠕动,缓释颗粒微丸会逐步进入空肠、回肠和结肠,崩解并释放药物产生治疗作用;市售制剂微丸在SD 大鼠胃肠消化道内流动性好,缓释颗粒微丸会较快进入空肠、回肠和结肠,崩解并释放药物产生治疗作用,同时,包衣在胃肠消化液中可能存在一定耐受性问题,使不同时间点胃肠消化道不同部位出现部分微丸的崩解,造成药物提前释放。
从美沙拉嗪不同部位药物含量看,自研制剂口服给药后在2~8 h 时间段胃部内容物均存在高浓度分布,并随着时间的推移较快进入并存在于空肠、回肠和结肠,随后在6~12 h 后,逐步在结肠达到高浓度分布,其结果有助于美沙拉嗪药物的体内吸收,以及缓释颗粒剂在作用部位的定点释放而发挥治疗效果;市售制剂SD 大鼠口服后,缓释颗粒微丸会较快进入十二指肠、空肠、回肠、甚至结肠,在2~6 h 时间段回肠内容物相对维持较高浓度分布,并随着时间的推移较快进入结肠,使结肠内容物中药物含量在4~12 h 时间段内持续较高水平,有利于药物在作用位点及时释放以提高疗效。
猜你喜欢
分子催化(2022年1期)2022-11-02
中国典型病例大全(2022年9期)2022-04-19
中国药学药品知识仓库(2022年5期)2022-04-11
现代牧业(2021年4期)2021-04-14
烟台大学学报(自然科学与工程版)(2021年1期)2021-03-19
微型计算机(2020年1期)2020-04-15
中国中药杂志(2016年23期)2017-04-07
中国中药杂志(2016年22期)2017-02-13
中国美容医学(2016年6期)2016-08-31
微型计算机(2009年4期)2009-12-23
