
沥青质微观聚集特征的分子动力学研究
2021-08-03 06:46黄世军赵凤兰赵大鹏
油气地质与采收率 2021年4期
王 鹏,黄世军,赵凤兰,赵大鹏
(1.中国石油大学(北京)石油工程教育部重点实验室,北京 102249;2.中国石油大学(北京)石油工程学院,北京 102249)
近几十年来,沥青质沉积问题给石油生产、运输和炼油等领域带来了许多工业挑战[1]。沥青质沉积不仅会造成储层孔喉堵塞,还会导致井筒和生产设备腐蚀结垢,严重影响油田的正常生产[2-3]。因此,油田开发过程中沥青质沉积的防治问题亟需解决。
沥青质结垢的清除成本高昂,如何规避或抑制沥青质沉积成为研究者们致力去寻找的解决方案。建立沥青质沉积的热力学预测模型是研究和预防沥青质沉积的重要手段之一,而对沥青质宏观尺度热力学行为准确建模的前提是需要了解其微观尺度的聚集机理,这需要明确沥青质分子聚集涉及的分子间作用力[4-5]。然而,由于沥青质的分子结构和官能团尚未准确测定,沥青质没有精确的定义和统一的分子构型。沥青质是石油中极性最强的重质组分,目前应用最广泛的是根据溶解度将其定义为可溶于芳族溶剂(如甲苯)而又不溶于正构烷烃(如正庚烷)的化合物[6]。溶解度定义中提及的沥青质一般特征不足以反映沥青质分子聚集涉及的分子间相互作用,关键还是要获取具有代表性的沥青质分子构型。
随着计算科学理论和计算机技术的进步,分子动力学方法得到广泛的应用。分子动力学方法是在分子和原子尺度上描述体系结构与行为的计算机模拟方法,不仅可以用来预测物质的性质、材料微观结构以及微观作用机理,还可以解决时间和成本的问题,具有物理模拟实验和理论计算不可比拟的优势[7]。丁勇杰依据三组分分离法,选择基于核磁共振分析和红外分析的两种沥青质分子构型,采用分子动力学模拟方法,探究沥青的化学结构特征[8];HEADEN 等开展经典原子分子动力学模拟研究,对比4种沥青质分子构型、胶质分子构型及其混合物在甲苯或正庚烷中的聚集特征[9]。沥青质分子结构对其聚集特征的影响较为显著,且沥青质分子结构多样,每一个研究案例都需要单独进行建模。因此,有必要建立一个具有代表性的沥青质平均分子构型。为此,采用分子动力学方法,结合沥青质分子单体代表性结构式、平均相对分子质量和分子基团特征,建立沥青质平均分子构型,并将其应用到沥青质模拟体系初始构型的构建,通过几何构型优化和退火处理后,模拟沥青质在地层环境中的运移过程,探究沥青质分子性质和微观聚集特征,进一步揭示沥青质沉积的微观作用机理。
1 模拟方法
1.1 沥青质模拟体系的分子动力学实验原理
分子动力学方法是基于经典力学原理,通过分子体系总势能求解系统中所有原子核的牛顿运动方程数值解,分析原子核位置和速度随时间的演化,来计算系统的动力学和热力学性质[10]。量子力学方法从原子的电子结构和分子的运动出发,通过求解不同亚原子粒子的波函数来描述粒子行为,是最精确的分子势能计算方法[7]。但即使是很小的系统,量子力学计算也非常昂贵,且超出了当前大多数计算机的计算能力。因此,研究者们建立了原子力场模型来近似计算分子势能[10]。力场方法是在经典力学的理论基础上,借助经验和半经验参数计算分子结构和能量的方法,它通过提供一组基于粒子相对位置的势函数和力学参数来计算系统的势能。
现有较为成熟的力场模型主要依据元素种类或原子所在官能团种类对分子中的各原子赋予不同的力场参数,计算分子内和分子间的相互作用,进而研究分子性质[11]。在力场模型中,化学键或所在官能团不同的同种元素会看作是不同种类的原子,这里的种类并非指真实的原子种类,而是力场模型中表征不同原子特征的分类,如烷烃上的碳、苯环中的碳和卟啉中的碳在力场模型中隶属于不同原子,会赋予不同的力场参数。本文研究的沥青质模拟体系,既包括有机大分子沥青质单体,又包括水和二氧化碳等无机分子,对于这样一个复杂的有机-无机复合系统,明确分子中原子的成键类型、官能团类型等结构特征,选择合适的力场模型赋予相应的力场参数,是保证沥青质模拟体系分子动力学模拟准确性的基础。
1.2 模拟体系初始构型的建立
采用Accelrys公司研发的Materials Studio 软件,建立沥青质平均分子构型,并通过Amorphous Cell模块组装沥青质模拟体系(图1)。Amorphous Cell是一种分子动力学无定形组装模块,能建立复杂无定型系统中的代表性模型,以特定密度组装多个分子,且各分子在模拟系统中的堆叠合理、仿真程度较高。以密度为1.2 g/cm3组装4 个沥青质分子、48个水分子和10 个CO2分子,模拟沥青质在地层中的运移环境,以二氧化硅分子层代表地层孔隙骨架,模拟盒尺寸为26.60 Å×9.83 Å×12.99 Å,在各方向上均采用周期性边界条件。

图1 地层环境下沥青质模拟体系的初始构型Fig.1 Initial configuration of asphaltene simulation system in formation environment
1.3 力场参数的确定
在进行分子动力学模拟实验前,首先需要选择合适的力场模型,并赋予相应的力场参数来计算粒子之间的相互作用,而力场参数的选择对分子动力学模拟结果的准确性至关重要。不同的力场模型适用的模拟体系有所不同,如AMBER(Assisted Model Building with Energy Refinement)力场主要适用于模拟生物大分子,而CHARMM(Chemistry at Harvard Macromolecular Mechanics)力场可以较好地拟合小分子体系和溶剂化的大分子体系。因此,针对地层环境下沥青质模拟体系的特点,从以下3 个方面考虑来确定力场参数。
沥青质分子的自聚集行为 沥青质作为原油中极性最强的组分,具有很强的自聚集倾向,不同浓度的沥青质分子的聚集形态有所不同[4]。当质量分数较低(小于10-4)时,沥青质以分散态的单分子形式存在;随着体系中沥青质质量分数的不断增加,沥青质分子开始发生聚集行为,聚集颗粒逐渐变大,依次形成纳米聚集体(通常含有8~10个沥青质分子)、纳米簇和黏弹性网络[12]。在选择与沥青质模拟体系相适应的力场模型时,首先要考虑的是能够较好地拟合沥青质这种有机大分子自聚集行为。
CVFF(Consistent Valence Force Field)力场可适用于计算各种多肽、蛋白质和大量的有机分子,在计算系统结构与结合能方面最为准确,可以较好地拟合沥青质分子自聚集行为和沥青质-胶质分子缔合行为[13]。其势能函数表达式为:

有机-无机复合体系 沥青质模拟体系中除了有机大分子沥青质,还包括水和二氧化碳等无机小分子,因此,需要选择一种能兼顾有机-无机复合体系的力场模型。COMPASS(Condensed-phase Optimized Molecular Potentials for Atomistic Simulation Studies)力场采用从头计算与经验方法相结合的方式计算力场参数,是第一个把有机分子体系的力场与无机分子体系的力场统一的力场模型[14]。其势能函数公式为:

(2)式中右侧前5 项为成键作用势能,后2 项为非成键作用势能,分别为静电相互作用和范德华力,其计算公式分别为:

分子力场的迁移性问题 力场参数是通过大量拟合基分子的实验数据得到的,若拟合的实验数据是在常温常压条件下测定的,而模拟的沥青质在地层中的运移环境往往是高温高压条件,此时应用这些力场参数时分子力学的准确性会降低[15]。Dreiding 力场是一种比较特殊的力场计算方法,可以计算一些缺乏实验数据的新结构化合物,是典型的经验力场模型。通过借助Dreiding 力场中的经验参数进行辅助参考,有助于提高分子动力学模拟实验结果的准确性。
1.4 沥青质分子动力学模拟的实验方法
沥青质分子动力学模拟的实验方法主要包括以下4 个步骤:①基于本文建立的沥青质模拟体系初始构型,选择合适的力场参数来计算系统中粒子间的相互作用。②采用最陡下降法调整偏离平衡位置较大的结构,再基于共轭梯度法对接近平衡状态的结构进行优化,使系统内分子达到能量最小值和原子位置最佳分布,实现沥青质模拟体系的几何构型优化。③在高温条件(303.15~503.15 K)下进行8个循环的退火处理,消除残余应力,退火过程持续8 ps。④采用玻尔兹曼分布函数,随机赋予系统中各分子运动的初始速度(图2),使用Forcite 模块在NVT 系综下模拟沥青质在地层岩石孔隙中的运移过程,模拟地层温度设为328.15 K,控温方法选择Nosé-Hoover 算法,时间步长为1 fs,模拟时长为15 000 ps。

图2 系统中各分子运动初始速度的玻尔兹曼分布Fig.2 Boltzmann distribution of initial velocity of each molecule in system
2 结果与讨论
2.1 沥青质平均分子构型的构建
沥青质分子的主体由含有若干个芳环组成的片状单元结构所构成,这些片状单元结构的个体间结构差异较大,根据芳核特征可将沥青质分子结构分为大陆型和群岛型[16]。由不同地区原油中沥青质的分子构型(图3)可以看出,不同地区的沥青质分子结构存在差异,如辽河和华北原油中的沥青质分子的脂肪族支链较长,塔里木原油中的沥青质分子则表现为标准的大陆型结构,而伊朗和俄罗斯原油中沥青质分子的芳环数量较多[17]。虽然不同地区的沥青质分子结构存在差异,但总体上看其结构特征也具有一般性,这为建立代表性的沥青质平均分子构型提供了参考。

图3 不同地区原油中的沥青质分子构型Fig.3 Molecular configurations of asphaltenes in crude oil samples from different regions
目前,已知的大多数沥青质分子单体结构包括1 个由4~10 个芳环组成的芳核和周围长度为3~7个碳原子的脂肪族链,这为构建沥青质平均分子构型的芳环数和支链长提供了参考依据[4]。ROGEL提出的一种基于沉淀法测定的委内瑞拉油样沥青质馏分的平均结构模型[18]被后续研究者沿用较多,其结构式具有一定的代表性,是本文沥青质平均分子构型构建的基础。而平均相对分子质量会影响沥青质的密度和溶解度等物理性质,可以作为构建沥青质平均分子构型的约束条件。随着测定仪器精度的提高和测量方法的改进,沥青质平均相对分子质量的测定值发生了几个数量级的变化,从最初测定的103~106到现在普遍认可的750[19]。综合多种沥青质分子结构式、平均相对分子质量(750)和沥青质分子基团结构的一般特征,构建出如图4 所示的沥青质平均分子构型,该分子构型的H/C 值为1.2,而沥青质分子的H/C 值一般为1.0~1.5,因此其结构具有一定的代表性。

图4 沥青质平均分子构型Fig.4 Average molecular configuration of asphaltenes
2.2 沥青质平均分子构型优化与退火处理
利用商业分子设计软件构建分子构型的原理往往不是第一性原理,而是依靠分子内固有的化学键的键长和键角,但这些数据往往是通过大量实验测得的平均数据,因此构建的初始分子构型往往不是目标化合物的稳定结构。为了得到更加真实的分子几何构象,在分子动力学中通常采用能量极小化方法,即在分子间作用力处于最小的状态下,调整分子几何构象使分子键长和键角接近“自然”状态,进而得到原子位置的最佳排布[10]。
从能量优化结果(图5)可以看出,系统能量总体上随着优化时间步数的增加而降低,但变化过程中存在明显的转折点。其原因是:由于势能面具有波动性,数学上只能保证求得局部极小值,需要采用多种构象搜索方法结合使用,来尽可能地达到全局最小值[20]。本文首先采用最陡下降法,其原理是根据能量梯度负值进行一维搜索,是能量极小化初期最简单、快捷的搜索算法,适用于分子结构偏离平衡位置较远[10]。但随着不断逼近极小值,其搜索方向总是垂直于上一步的特性使得搜索到的势能值总是在极小值附近震荡而无法收敛,因而可以看到优化时间步数为0~380时能量衰减趋势缓慢且波动频繁,此时需要采用更加精细的共轭梯度算法,通过不断迭代,快速逼近系统能量的全局最小值,该过程对应于优化时间步数为380~500时系统能量衰减速度显著增加,直到达到最低势能构象,即最稳定的构象。
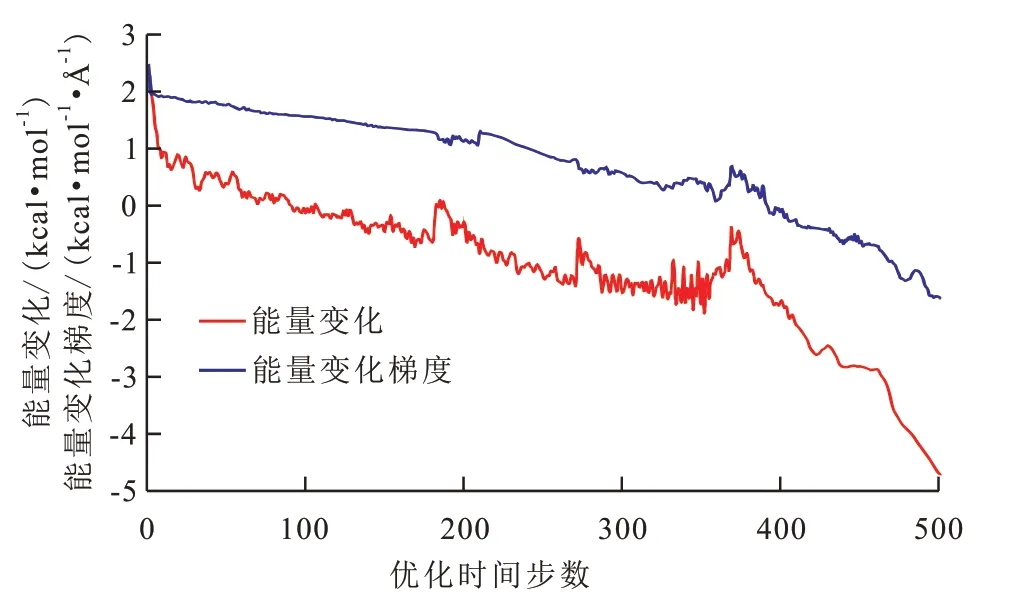
图5 沥青质模拟体系初始构型能量优化Fig.5 Energy optimization of initial configuration of asphaltene simulation system
对沥青质模拟体系初始构型进行几何构型优化后,完成系统温度从303.15 K 到503.15 K 的8 个循环退火处理,退火过程中系统的各种能量变化如图6 所示。由于退火过程中系统温度波动,总动能和总势能的变化也存在一定波动,但相比于初始构型,随着系统温度升高,分子热运动加剧,系统总动能和总势能增加。随着分子间距增加,分子间的范德华力相互作用减弱,静电相互作用将占据系统非成键作用中的主导地位。在高温条件下,沥青质分子内各原子热运动加剧,能够充分暴露其内部结构,随后通过退火处理,系统温度缓缓降低,在每个温度都达到结构的平衡态,如此循环往复退火过程,消除残余应力,使沥青质分子结构达到能量基态。

图6 退火过程中系统能量变化Fig.6 Energy change during annealing
2.3 沥青质分子构型的径向分布特性
径向分布函数是表征粒子微观结构特征的物理量,其物理意义为任意指定的中心粒子周围距离为r处出现其他粒子的概率,如图7所示。它反映了凝聚态物质粒子聚集的径向有序性,其特点为“以宏表微”,即只需测定沥青质的宏观数值就可以得到其微观细节[21-23]。

图7 径向分布函数概率分布示意Fig.7 Probability distribution of radial distribution function
径向分布函数可以理解为系统的局部密度与平均体密度的比值,其计算式[23]为:

分子动力学模拟获得的系统各种统计学平均性质,主要通过统计分析函数来计算轨迹文件中关于各种性质的数据并绘制分布图,如总能量、总势能、总动能、速度和密度等随时间的变化。g(r)同样也可以从轨迹数据文件中获得,其实质就是原子间矢量长度的球型平均分布[24]。对非晶体而言,当r值很大时,g(r)的函数值趋近于1,表明距离参考粒子为r的位置的粒子分布无规律性;而当r值相对较小时,g(r)的函数值会在小范围内出现几个波峰(极大值),即这些位置处的局部密度与平均体密度的比值较大,说明波峰位置处出现粒子的概率要明显高于其他位置。总体上,非晶体的径向分布特征表现为近程有序、远程无序[25]。
从沥青质平均分子构型的径向分布函数(图8)可以看出,g(r)总体上呈现近程波动剧烈且分散、远程波动平缓且密集的特征。当r<0.25 nm 时,g(r)表现出较强的规律性,分别在0.11,0.14,0.17 和0.22 nm等处出现波峰,这些波峰表明从参考粒子到该位置处出现粒子的概率要远高于其他位置,主峰位置一般标志着聚集结构的出现,这可能与沥青质分子的稠环结构有关;而当r>0.5 nm时,g(r)的函数值趋近于常数1,说明粒子的分布没有规律,呈无序状态。沥青质是典型的非晶体,在成键作用下分子内各原子呈有序排列状态,表现为近程有序;随着r值增大,沥青质的分子聚集状态方位会对沥青质的出现频率产生影响,在常见的方位配置中,具有非面对面配置方式的沥青质分子径向分布规律性较差,其出现频率无规则,分子堆积更松散,因此远程表现为无序状态。

图8 沥青质平均分子构型的径向分布函数Fig.8 Radial distribution function of average molecular configuration of asphaltenes
2.4 沥青质在地层岩石中的运移模拟
沥青质在地层环境运移过程中,系统内各分子运动的初始速度由玻尔兹曼分布函数随机产生,模拟过程中系统温度在设定值328.15 K上下波动。从沥青质模拟体系在地层中的运移终态(图9)可以看出:大量水分子向模拟盒的两端运移,并最终倾向于吸附到二氧化硅分子层表面,这验证了二氧化硅的强亲水性;而二氧化碳分子在整个分子动力学模拟过程中位移量较小,这可能与其是非极性分子有关,且目前文献中也尚未有记载关于从分子间的相互作用的角度去解释二氧化碳对沥青质沉积的直接影响;沥青质分子中的芳核主体以及部分脂肪族链均不同程度的向二氧化硅分子层表面靠近,并倾向于逐渐被吸附到二氧化硅分子层表面,沥青质分子单体或聚集体在二氧化硅表面的吸附造成孔隙内壁的非均质性会改变接触角,是导致孔隙润湿性改变最可能的原因之一,这一点在ANDREW 等[26]和LI 等[27]的实验研究中都得到了验证。岩石表面的润湿性由亲水性向亲油性转变,是沥青质沉积导致原油采收率降低的重要机理之一。
此外,从图9 中亦可观察到沥青质分子间的聚集状态,聚集的驱动力来自于沥青质分子中芳核的π-π 堆积作用,而脂肪族支链引起的位阻则是沥青质分子聚集的阻力。π-π 堆积作用是由于芳香族环的负π 电子云和正σ框架之间的静电吸引而产生的,常见的堆积方式包括面对面或平行、错位堆叠或平行错位、以及边对面或T形[4]。其中,图10中所示的堆积方式为面对面堆积,是最稳定的苯二聚体构型,它能够最大程度地减少负π 电子云之间的相互排斥作用。沥青质分子中芳核发生的π-π 堆积作用引发了沥青质分子聚集,最终导致沥青质絮凝并从原油中沉积出来,堵塞岩石孔喉并导致原油采收率降低。

图9 沥青质模拟体系运移终态Fig.9 Final migration state of asphaltene simulation system

图10 沥青质分子二聚体构型示意Fig.10 Configuration of asphaltene molecular dimer
3 结论
基于分子动力学方法,结合多种沥青质分子结构式、平均相对分子质量和沥青质分子基团结构的一般特征,建立了沥青质平均分子构型。该构型具有一定的代表性,并用于构建沥青质模拟体系初始构型来模拟沥青质在地层岩石中的运移。
沥青质作为典型的非晶体,其径向分布特征总体表现为近程有序、远程无序。当周围概率粒子到参考粒子的距离较小时,在成键作用下分子内各原子呈有序排列状态;随着周围概率粒子到参考粒子的距离增大,具有非面对面配置方式的沥青质分子径向分布规律性较差,分子堆积更松散。退火处理过程中系统的各种能量变化表明,退火优化可使分子链充分松弛,有利于模拟该分子结构的能量初态。随着分子间距增加,分子间的范德华力相互作用减弱,静电相互作用将占据系统非成键作用中的主导地位。
从沥青质模拟体系的运移终态可以看出,沥青质分子的芳核主体及部分脂肪族链倾向于逐渐被吸附到二氧化硅分子层表面,这会使岩石表面的润湿性由亲水性向亲油性转变;沥青质芳核的π-π 堆积作用是引发沥青质分子聚集的驱动力,从而导致沥青质絮凝并从原油中沉积出来。润湿性反转和岩石孔喉堵塞是沥青质沉积造成原油采收率降低的重要机理,明确沥青质分子的微观聚集特征,能够有效预防原油开采过程中潜在的储层伤害风险,尤其是在注气提高采收率技术应用过程中。
符号解释
a,Dr——键伸缩能计算参数;
Aij——范德华相互作用参数,代表原子间的排斥作用;
b,b0——分子的键长和平衡键长,nm;
Bij——范德华相互作用参数,代表原子间的吸引作用;
dN——球形壳内的粒子数目,个;
dr——球形壳的厚度,nm;
g(r)——径向分布函数;
i,j——体系中任意两个不同原子;
kθ——键角弯曲能计算参数;
Kϕ——键扭转能计算参数;
kx——键角面外弯曲能计算参数;
n——键扭转能、键角面外弯曲能参数;
qi——原子i的电荷量,C;
qj——原子j的电荷量,C;
r——周围概率粒子到参考粒子的距离,nm;
rij——原子i与原子j之间的相对距离,nm;
——原子i与原子j间范德华势能为0时的距离,nm;
U——总势能,kJ/mol;
Ub——键伸缩能,kJ/mol;
Ucross——耦合能,kJ/mol;
Ux——键角面外弯曲能,kJ/mol;
Uθ——键角弯曲能,kJ/mol;
Uϕ——键扭转能,kJ/mol;
Ucoul——静电势能,kJ/mol;
Uvdw——范德华势能,kJ/mol;
x,x0——分子的离平面振动角度、平均离平面振动角度;
θ,θ0——分子的键角、平衡键角;
ϕ,ϕ0——分子的二面角、平衡二面角;
ρ——密度,g/cm3;
ε0——介电常数;
εij——势阱深度。
猜你喜欢
军民两用技术与产品(2022年1期)2022-06-01
西北工业大学学报(2022年1期)2022-04-22
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
房地产导刊(2021年8期)2021-10-13
天津理工大学学报(2021年2期)2021-06-03
中学生数理化(高中版.高考理化)(2021年12期)2021-03-08
高中生学习·高三版(2017年9期)2017-10-26
中学生数理化·八年级物理人教版(2016年5期)2016-08-26
