
新城疫强毒特异性反转录荧光定量PCR检测方法的建立与应用
2021-09-12 09:44徐敏丽于江逯璐张玉玉任素芳陈智孙文博郭立辉吴家强张琳
山东农业科学 2021年8期
徐敏丽,于江,逯璐,张玉玉,任素芳,陈智,孙文博,郭立辉,吴家强,张琳
(1.山东省农业科学院畜牧兽医研究所/山东省畜禽疫病防治与繁育重点实验室,山东 济南 250100;2.山东管理学院新兴业态发展研究所,山东 济南 250357)
新城疫(newcastle disease,ND)是由新城疫病毒(Newcastle disease virus,NDV)引起的一种急性、高度接触性的禽类传染病,是世界动物卫生组织(OIE)规定的法定报告疫病,给许多国家养禽业造成巨大经济损失[1]。NDV又称禽副黏病毒1型,在分类上属于单链负股病毒目副黏病毒科副黏病毒亚科的禽腮腺炎病毒属,其基因组包含6个开放阅读框,从5′到3′依次编码L-HN-FM-P-NP共6种结构蛋白[2,3],其中F蛋白是最主要的糖蛋白之一。病毒毒力的强弱与其裂解位点区的氨基酸序列相关,强毒株氨基酸序列一般为112R/K-R-Q-K/R-R-F117,弱毒株则为112G/E-K/R-Q-G/E-R-L117,氨基酸序列上的差异也成为在基因水平上区分NDV强、弱毒株的靶点。
目前,我国对于新城疫的防控主要依靠疫苗,有效的疫苗和合理的免疫程序很大程度上控制了该病的暴发和流行。但由于我国养殖环境复杂,标准化、规模化养殖水平不一,致使NDV在养殖环境中长期存在[4],这也给病毒的变异进化提供了条件,不但在宿主范围和致病性上产生了变化,而且常以非典型病症呈现[5-7],所以NDV的鉴定和病例的确诊必须经过实验室诊断。当前,NDV检测主要使用PCR技术和血清学技术。相比于血清学技术,PCR技术在病毒的快速检测中具有先天性优势,不仅耗时短、操作简便,而且敏感性更高。然而,由于疫苗(灭活疫苗和弱毒苗)的广泛使用和一些天然弱毒株的存在[8],从免疫禽类中检测到的NDV可能是被接种的疫苗病毒或是不至于引起发病的弱毒株,即使检测和分离到了NDV,也不能确定禽类感染了野毒。所以,准确掌握NDV的感染情况,首先要排除疫苗毒的干扰,能够快速、准确区分NDV强、弱毒株或者特异性扩增NDV强毒的检测方法十分必要。
对于区分NDV强、弱毒株的PCR检测方法已有报道,曹殿军[9]、胡传伟[10]、黄淑坚[11]等设计了两对特异性引物分别扩增NDV强、弱毒株,但所建方法的灵敏度不高;岳华[8]、曹军平[12]、王珂[13]等分别建立了基于探针法的针对NDV强毒的荧光定量RT-PCR方法,在检测灵敏度方面有很大程度的提升,但探针的应用使得检测成本大幅提升,在广泛应用中受到限制。为此,急需一种低成本、高敏感性的NDV强毒特异性荧光定量PCR检测方法。本研究基于荧光定量PCR高效、灵敏的优点,以SYBR GreenⅠ染料代替探针,配以特异性引物,建立了基于SYBR GreenⅠ嵌合荧光的NDV强毒特异性逆转录荧光定量PCR(reverse transcription fluorescence quantitative PCR,RT-qPCR)检测体系,能够为新城疫的监测提供预警信息,从而有效控制NDV强毒感染的发生与蔓延。
1 材料与方法
1.1 毒株
NDV强毒(F48E9)、NDV弱毒(lasota、克隆30、V4)、禽流感病毒(Avian influenza virus,AIV)、禽呼肠孤病毒(Avian reovirus,ARV)、鸡传染性支气管炎病毒(Infectious bronchitis,IBV)、鸭病毒性肝炎病毒(Duck hepatitis A virus,DHAV)、鸡传染性喉气管炎病毒(Infectious laryngotracheitis,ILTV)、禽坦布苏病毒(Avian tembusu virus,ATMUV),均由山东省畜禽疫病防治与繁育重点实验室分离鉴定并保存。
1.2 试剂
RNAiso Plus、T7 RNA Polymerase、One Step RT-PCR Kit、TB Green PremixEx Taq嵌合荧光法检测试剂盒、Ex TaqHS,均购自大连宝生物科技有限公司;Simply P病毒DNA/RNA提取试剂盒,杭州博日科技股份有限公司。
1.3 试验方法
1.3.1 引物设计 根据GenBank中登录的所有NDVF基因序列,分析强、弱毒区分位点,设计强毒特异性引物对,以达到只扩增强毒而不扩增弱毒的目的。上下游引物序列如下:NDV F realtime-1:5′-TACACCTCATCCCAGACAGG-3′,NDV F realtime-2:5′-AGTCGGAGGATGTTGGCAGC-3′,扩增产物长度为305 bp。另设计合成含T7启动子的上游序列,NDV F realtime-T7-1:5′-TAATACGACTCACTATAGGGTACACCTCATCCCA GACAGG-3′,用于体外转录模板的制备。
1.3.2 核酸提取和体外转录模板的制备 提取NDV、AIV、ARV、IBV、DHAV、ILTV和ATMUV的核酸,保存于-80℃备用。以F48E9的RNA为模板,以NDV Frealtime-T7-1、NDV Frealtime-2为引物,经一步法RT-PCR扩增得到的目的条带作为体外转录模板。一步法RT-PCR反应体系为:PrimeScript one step enzyme mix 2μL,2×one step buffer 25μL,NDV F realtime-T7-1引物(100 pmol/μL)1μL,NDV F realtime-2引物(100 pmol/μL)1μL,RNA模板2μL,RNase Free Water补足50μL。一步法RT-PCR反应条件为:50℃反转录30 min;95℃预变性2 min;94℃变性1 min,55℃退火30 s,72℃延伸1 min,共32个循环;72℃延伸10 min。
1.3.3 NDV强毒RNA标准品的制备 取1μL体外转录模板进行体外转录,操作按T7 RNA Polymerase试剂盒说明书进行。产物最终溶解于DEPC水中,并用紫外分光光度计测其浓度,通过公式将得到的RNA换算为拷贝数,并用RNase Free Water进行10倍倍比稀释,将稀释的RNA作为标准品,-80℃保存备用。
1.3.4 RT-qPCR反应体系及标准曲线的建立 配制RT-qPCR反应体系并进行引物浓度、退火温度等条件的优化。反应体系(20μL):2×One Step SYBR RT-PCR buffer 10μL,Ex TaqHS 0.4μL,Primescript RT enzyme mixⅡ0.4μL,NDV F realtime-1、NDV F realtime-2引物(10~50μmol/L)各1μL,RNase Free dH2O 5.2μL,total RNA 2μL。反应条件:42℃反转录5 min;95℃预变性3 min;94℃变性10 s,53~60℃退火10 s,72℃延伸25 s,共40个循环;72℃延伸10 min。其中引物浓度取10、20、30、40、50μmol/L共5个浓度梯度;退火温度选择53、55、57、58、60℃,每个反应条件均进行3次重复。结果根据反应的扩增曲线和溶解曲线进行判定。随后根据反应体系和条件,进行标准曲线的建立,选择1×108、1×107、1×106、1×105、1×104、1×103拷贝/μL 6个浓度梯度的RNA标准品作为模板进行RTqPCR扩增,每个浓度的标准品3个重复,扩增后用于标准曲线的建立。
1.3.5 RT-qPCR方法的敏感性 分别以1×106、1×105、1×104、1×103、1×102、1×101、1×100拷贝/μL的RNA标准品作为模板,使用优化的RT-qPCR反应体系进行扩增,能够有效扩增的最低稀释度的拷贝数为检测方法的检测极值。
1.3.6 RT-qPCR方法的特异性 采用优化的体系和条件,以NDV强、弱毒和其他禽类常见病毒(AIV、ARV、IBV、DHAV、ILTV和ATMUV)的核酸为模板进行RT-qPCR检测,以确定该方法的特异性。
1.3.7 RT-qPCR方法的准确性 利用建立的RT-qPCR方法对本实验室保存的NDV阳性样本(表1,已测定F基因全序列)进行准确性验证。经RT-qPCR方法扩增,与测序结果相比较以验证RT-qPCR方法的准确性。

表1 NDV阳性样本
2 结果与分析
2.1 体外转录模板和RNA标准品的制备
以NDV F realtime-T7-1和NDV F realtime-2为引物,F48E9 RNA为模板进行体外转录模板的制备。经琼脂糖凝胶电泳和序列测定显示,成功获得F48E9的目的基因片段。分光光度计测得其浓度为3.2×109拷贝/μL,对其进行10倍系列稀释后,用于标准曲线的建立。
2.2 RT-qPCR反应体系和标准曲线的建立
RT-qPCR反应体系的建立需要在常规体系的基础上进行条件优化。将引物浓度和退火温度进行正交试验,结果显示,当引物浓度为10 μmol/L、退火温度为55℃时体系的扩增效率明显优于其他条件(图1),且在该反应条件下溶解曲线单一(图2),确定为反应体系的最优条件。根据反应最佳条件,分别以1×108、1×107、1×106、1×105、1×104、1×103拷贝/μL浓度梯度的RNA标准品为模板进行RT-qPCR反应,获取各自的动力学曲线(图3),各浓度间重复性良好,以此获得的标准曲线方程为Y=-3.307X+2.657(图4)。
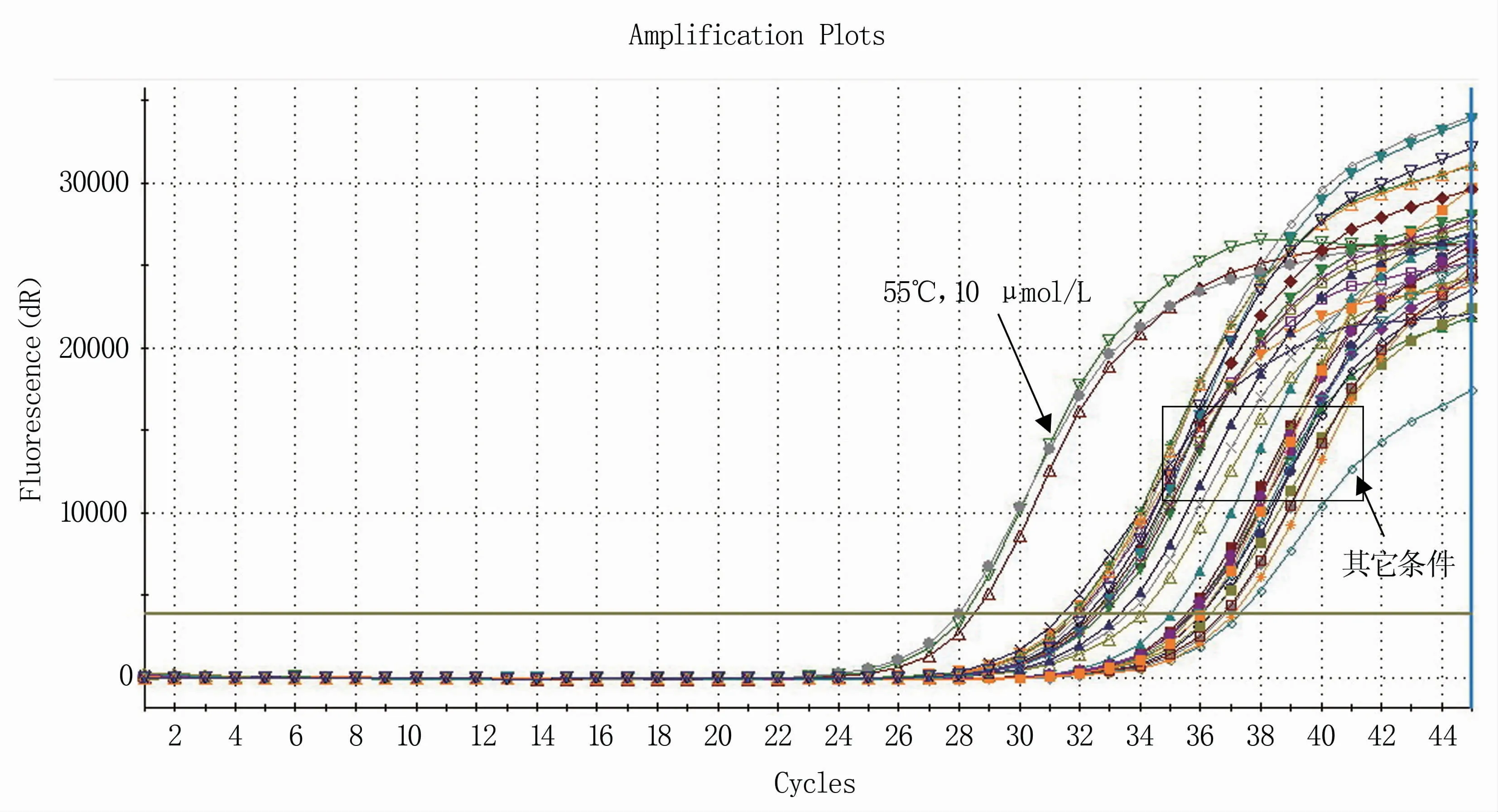
图1 不同反应条件下的扩增结果
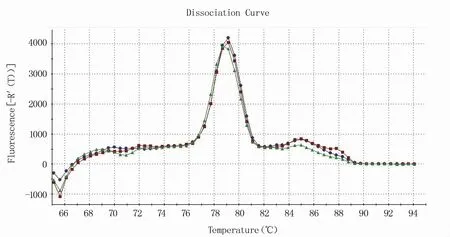
图2 最佳反应条件下的溶解曲线
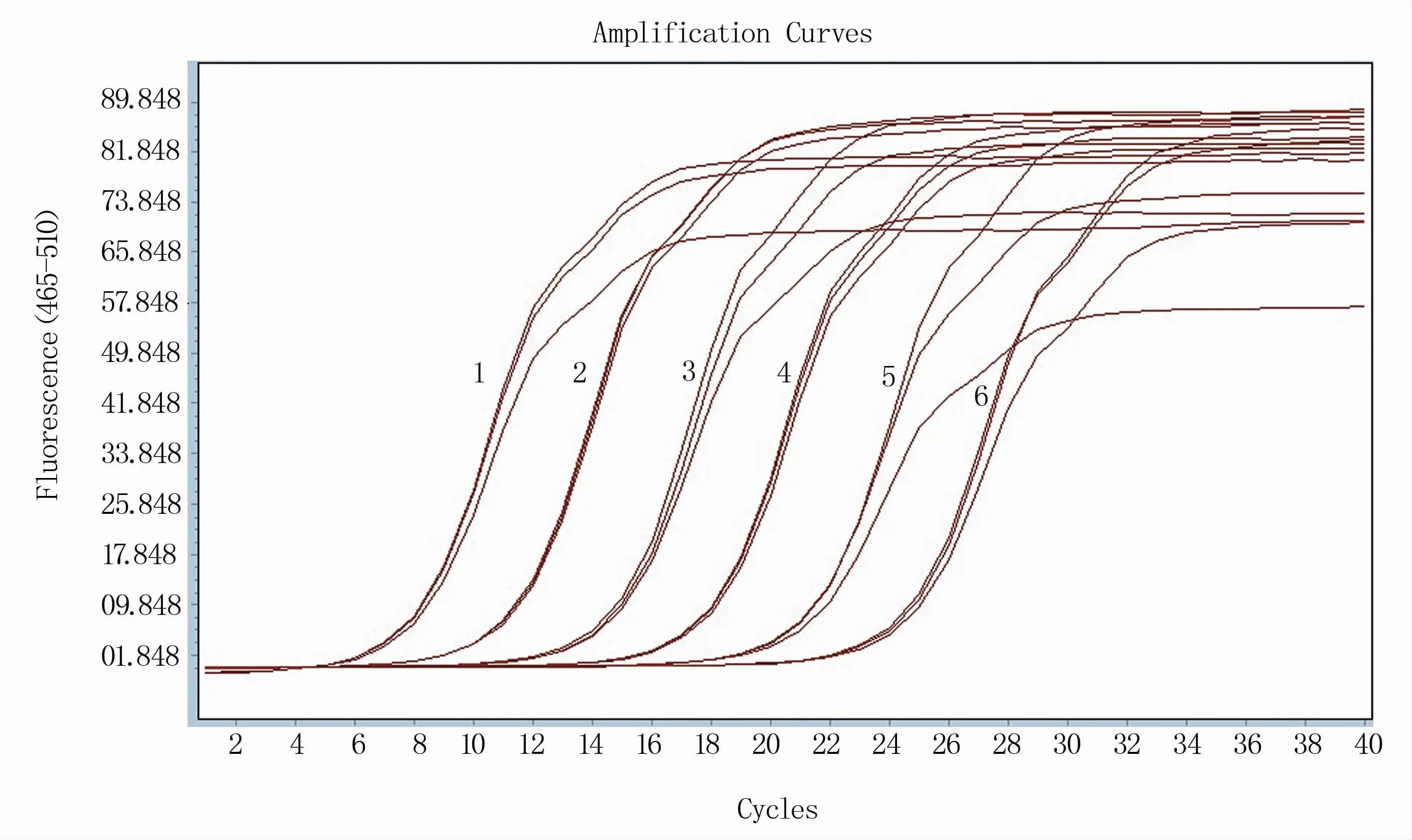
图3 不同浓度标准品扩增结果
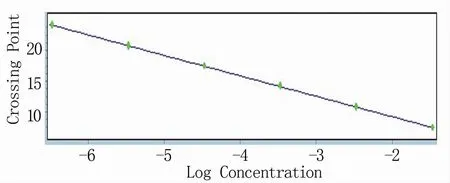
图4 标准曲线
2.3 RT-qPCR方法的敏感性
如图5所示,RT-qPCR能检测出的模板最低浓度为1×100拷贝/μL,且阴性对照无扩增,表明该方法的检测极限值为1拷贝/μL。
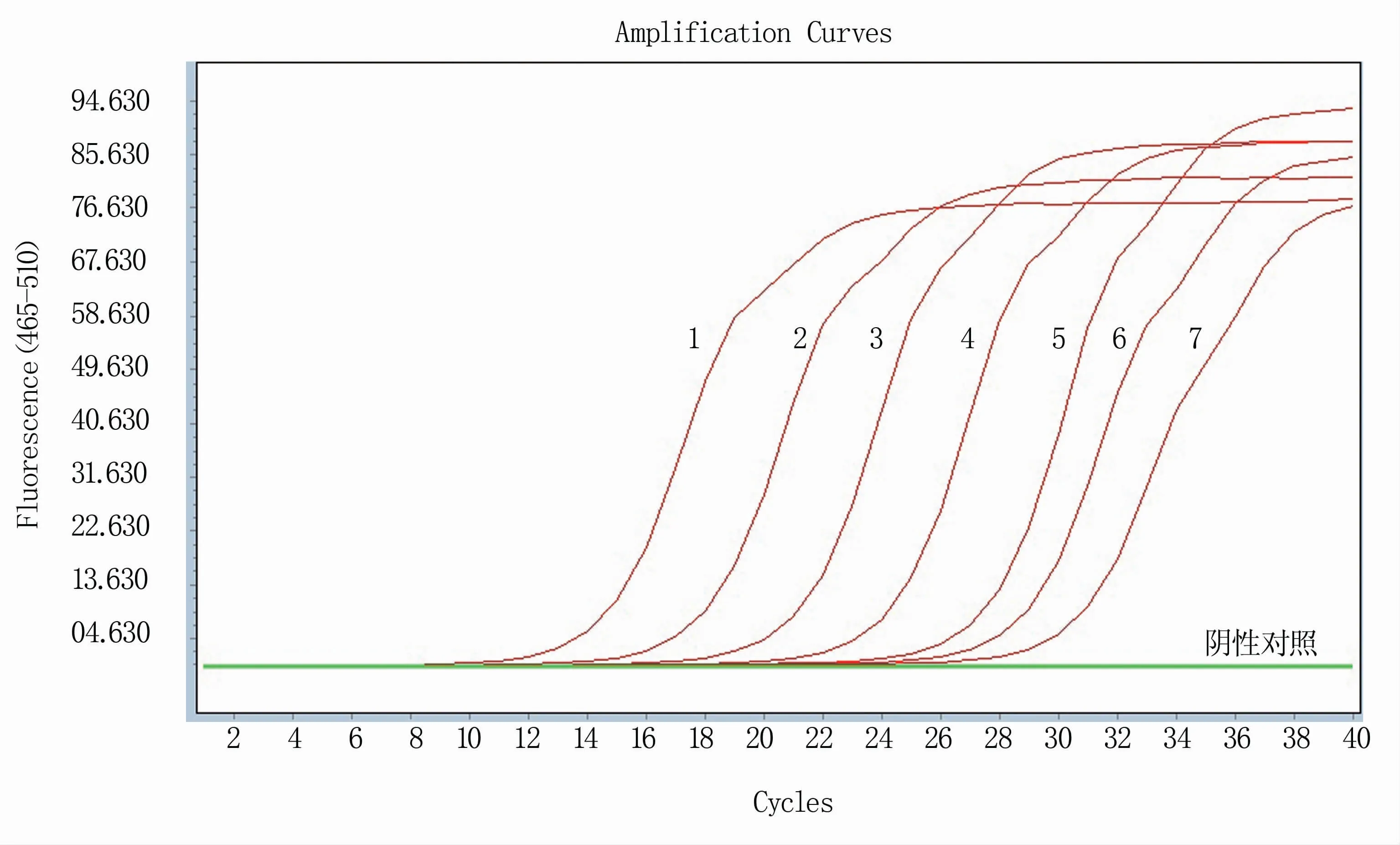
图5 RT-qPCR方法敏感性试验结果
2.4 RT-qPCR方法的特异性
应用建立的RT-qPCR方法对NDV强毒(F48E9)、NDV弱毒(lasota、克隆30、V4)、AIV、ARV、IBV、DHAV、ILTV和ATMUV进行检测,结果(图6)显示,仅NDV强毒(F48E9)成功被扩增,而NDV弱毒(lasota、克隆30、V4)、AIV、ARV(Ct值>35)、IBV、DHAV、ILTV、ATMUV则没有被扩增出。
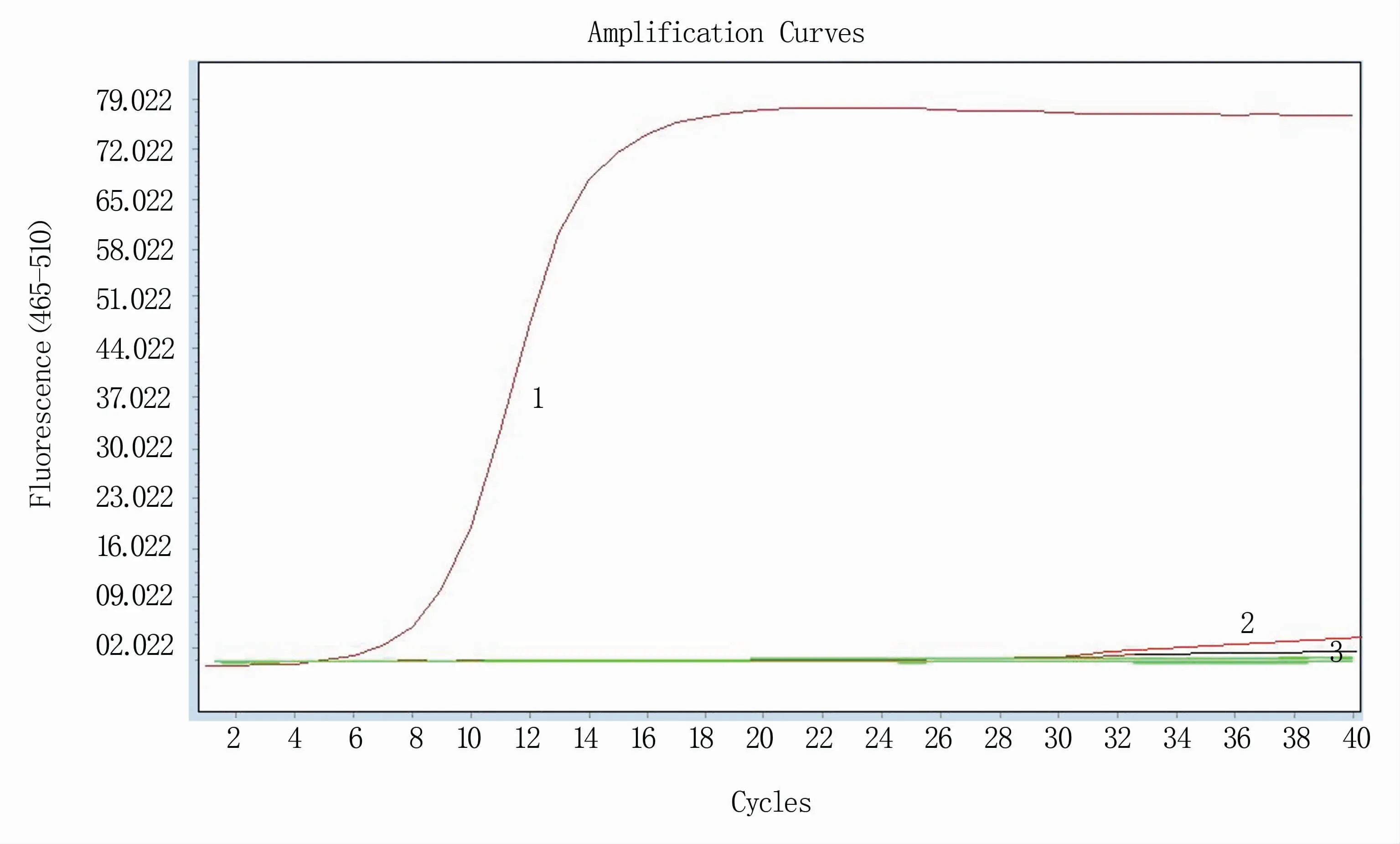
图6 RT-qPCR方法特异性试验结果
2.5 RT-qPCR方法的准确性验证
利用建立的RT-qPCR方法对25份已明确毒力的病毒进行检测,结果(图7)显示,10株含强毒序列特征的毒株RNA均能有效扩增,而其余毒株RNA则无法扩增,说明该检测方法准确、可靠。
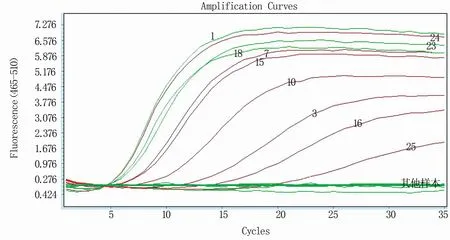
图7 RT-qPCR方法准确性验证
3 讨论与结论
NDV F蛋白不仅是介导病毒脂蛋白囊膜与宿主细胞表面融合的主要因子,还是决定病毒毒力的主要蛋白之一。F蛋白最初是以无活性F0的形式存在,在裂解成F1和F2之后,病毒才具有感染性,而F0的裂解能力取决于裂解区的氨基酸组成,如裂解区存在的碱性氨基酸越多,其裂解活性越强,反之则越弱。所以存在两对碱性氨基酸(精氨酸或赖氨酸)的病毒具有较强的裂解活性,其毒力也较强,而仅存在一对碱性氨基酸病毒的F0蛋白不易被裂解,从而降低了病毒囊膜与宿主细胞膜的融合活性,使病毒感染性降低或者无感染性[14,15]。研究表明,绝大多数的禽源NDV强、弱毒株均符合以上规律,所以根据F基因设计特异性引物用以区分NDV强、弱毒株也是较为普遍的方法。Nidzworski等[16]于2011年设计了一种区分NDV强、弱毒株的RT-PCR方法,但分析序列时发现其体系中的引物无法与后来分离的病毒特异性匹配,即病毒核酸的变异使得该检测方法失效。本研究引物的设计是基于对所有NDVF基因序列的分析,选择强毒与弱毒保守但有所区分的位点,能够保证较长的使用时效,结果也证实设计的引物能够特异性地扩增强毒RNA,而无法扩增弱毒RNA。
在NDV强毒特异性RT-qPCR检测方法建立的基础上努力提高敏感性是本研究的目标之一。一般情况下,NDV感染禽类3天后,典型症状才逐步显现,此阶段是病毒复制的对数期,更是控制病毒繁殖与扩散的关键期,如能及时检测到强毒株的存在并采取相应措施,能够更好地控制疫情的发生。所以,只有高敏感性的检测方法才能对早期感染做出准确判断。2000年,曹殿军等[9]设计的鸡NDV强、弱毒株RT-PCR鉴别诊断方法的敏感性仅为500 pg;黄淑坚等[11]设计了两对特异性引物,建立了区分NDV强、弱毒株的RTPCR方法,其敏感性为10 pg,换算成拷贝数大概为105拷贝,可见常规PCR的检测灵敏度无法满足现在的需求。2007年,岳华等[8]报道了基于探针法的NDV中强毒株荧光定量PCR检测方法,敏感性为60拷贝/μL,较之前有了极大的改善;2012年,检测体系的灵敏度达到了3拷贝/μL[12]。本研究所建立的NDV强毒特异性RT-qPCR检测方法的敏感性达到1拷贝/μL,能够更好地服务于NDV强毒的监测预警。
我国禽类养殖的基数大,且存栏量逐年上升,对于NDV强毒流行情况的大规模检测,控制成本是重要的环节。本研究使用SYBR GreenⅠ嵌合荧光显色技术,检测成本更低,更适合基层及大规模检测使用。在低成本下实现高敏感性是检测方法最终的追求,本研究建立的NDV强毒特异性RT-qPCR检测方法敏感性高、特异性强且成本较低,是NDV强毒监测更为合适的方法和工具。
猜你喜欢
动物医学进展(2022年9期)2022-11-26
现代仪器与医疗(2022年4期)2022-10-08
电气技术(2022年6期)2022-06-27
科学大观园(2022年2期)2022-01-23
文萃报·周二版(2021年47期)2021-12-14
现代临床医学(2021年1期)2021-01-26
记者观察·下旬刊(2019年12期)2019-09-10
今日中国(2017年8期)2017-09-03
今日中国·中文版(2017年8期)2017-08-14
诗歌月刊(2014年12期)2015-04-14
