
芳香性概念的最新进展
2021-09-26 10:50黄宝磊孙昊李欣烨范雨亭陶涛
大学化学 2021年8期
黄宝磊,孙昊,李欣烨,范雨亭,陶涛
南京信息工程大学化学与材料学院化学系,南京 210044
芳香性作为化学中最重要的概念之一,一直吸引着实验和理论科学家的关注。芳香性并不是一个严格意义上的科学概念,从未被完整定义,自从这一概念被提出以来,经历了150多年的发展,但这个古老的课题仍然有着鲜活的生命力,是有机化学中的前沿课题。作为一种概念,实验测定和量子化学的理论计算在对芳香性的评估中均展现出重要作用,特别是近几年,这一概念的内涵与外延不断扩展。尤其在新颖芳香性的发现和诠释中扮演着不可或缺的角色。
1 芳香性的研究历史
1825年,法拉第发现了苯,后来人们将一类具有芳香气味的化合物归为“芳香化合物”,但是由于这种归类具有主观性、模糊性,所以并未深入研究。1855年,霍夫曼[1](August Wilhelm Hofmann)最早使用aromatic一词表示含苯环的化合物,成为芳香性(aromaticity)的滥觞。1865年,德国有机化学家凯库勒(Friedrich August Kekulé, 1829–1896)提出苯的结构。1890年,凯库勒在德国化学会庆祝苯结构论文发表25周年的演讲中提到,他在梦中看到蛇咬着自己的尾巴,并受此启发,想到苯分子的环状结构。伴随着量子化学的发展,理论化学迎来了突破。1931年,德国科学家休克尔[2](Erick Hückel,1896–1980)提出具有(4n+ 2)个π电子的平面闭合共轭单环化合物可能具有芳香性,之后合成的一系列的芳香化合物也证明了休克尔规则的正确性。凯库勒1896年逝世,休克尔1896年出生,科学界将其称为“苯的连续之谜”(benzene enigma continues),以此表达对两位科学巨擘的敬仰。
1965年,Breslow[3]将(4n)个π电子的反芳香性补充到休克尔规则中。1972年,贝尔德(Colin Baird)提出当分子处于第一激发三重态时,其(反)芳香性与休克尔规则相反[4]。这两个经典规则在芳香性的理论体系中具有划时代的意义,指导了之后的众多相关研究。近些年来,除了传统的有机分子,金属苯及其衍生物、其他金属有机化合物的芳香性均有诸多报道,全局芳香性、三维芳香性、超共轭芳香性等新概念不断涌现(表1)。

表1 近年来有关芳香性化合物的研究进展
2 芳香性的判据发展
化合物是否具有芳香气味是判断芳香化合物的起源,但是这一判据过于主观粗糙,且不能反映这类化合物的物理、化学性质,所以逐渐被摒弃。虽然科学家对芳香性进行了深入研究并提出多种方法[12],但是最经典、被广泛认可的是休克尔规则,并沿用至今。1931年,休克尔等人提出:在单双键交替的平面环状结构中,具有(4n+ 2)个π电子的为芳香性;后又补充为具有(4n)个π电子的为反芳香性。1964年,Heilbronner预测具有莫比乌斯环状共轭拓扑结构的π体系中,具有(4n)个π电子的为芳香性,而不是(4n+ 2)个π电子[13],这与传统的休克尔规则刚好相反,称为莫比乌斯芳香性规则(Möbius aromaticity rule)。1972年,贝尔德提出不仅在莫比乌斯特殊的环状条件下,当分子处于三重态时亦存在与休克尔规则相反的情况。
如何判断化合物具有芳香性是芳香化合物被发现以来经久不衰的课题。芳香性是用来评价环状共轭有机化合物具有额外稳定性的一种概念,芳香性的形成是π电子良好重叠与π电子数合适两方面共同决定的,前者需要以σ键骨架作为结构基础,后者需要判断具体适用于休克尔规则还是贝尔德规则。下面从实验和理论两个方面进行举例阐述。
2.1 芳香性的实验判据
核磁共振谱(NMR)仍是判断芳香性的重要证据。苯的1H NMR谱图中,由于所有氢原子为磁等价和化学等价,化学位移值约为7.33,而同等条件下共轭烯烃的氢原子化学位移值为4.5–6.5,主要由于存在去屏蔽效应。[18]轮烯中,12个氢原子(12H)朝外,6个氢原子(6H)朝里。以氘代四氢呋喃为溶剂,−60 °C的1H NMR谱图中,12H化学位移值为9.25,6H化学位移值为−2.9。而在温度为120 °C的谱图中,所有的氢化学位移为5.45。这一实验结果表明:(1) 高温下,[18]轮烯朝外的氢与朝里的氢可以快速交换;(2) 芳香性作为一种理化性质,依赖温度的变化而变化。
除了利用休克尔规则或贝尔德规则以外,科学家曾试图通过芳环结构易取代难加成的化学反应特性、键长的平均化程度,以及几何构型等角度来判断芳香化合物,但是随着对芳香性研究得越深入,越显得这些判据较为粗浅。例如,富勒烯C60为足球状化合物,其易加成的性质突破了反应特性的限制。
此外,单晶结构测定获得分子的键长、键角等数据,也可以从实验上辅助判断分子的芳香性。键长平均化程度能够侧面反映共轭的情况,置信度较高,但是其平均化程度只可与同分异构体或同系物比较,才具有实际意义。例如,环硼氮烷(B3N3H6)中,所有B-N键键长均为0.143 nm[14],而萘分子并不是等键长体系,但是一般认为萘比环硼氮烷更具有芳香性。目前的实验判据,均为多种结果并存分析,只依赖一种实验结果就断定某种新体系是否具有芳香性,显得比较武断。
2.2 芳香性的理论判据
实验判据只能通过谱图间接表现,无法实现芳香性的可视化。但是随着理论计算和图形可视化技术的发展,这一状况得以改善。在芳香性体系中,离域的电子在外加磁场的作用下会产生明显的感应电流,因此研究分子的电磁性质是研究芳香性的有效手段。通过绘制矢量图,可以直观地观察到分子内感应电流,从而可以判断分子有无芳香性。其中,各向异性感应电流密度和核独立化学位移的优势逐步显现,成为主流的理论判据。
各向异性感应电流密度(anisotropy of the induced current density,AICD[15])本质上反映的是相应位置电子对磁场感应的各向异性的强度。其中,环电流方向与左手规则相同时,电流越大则芳香性越强;而电流方向越与左手规则相反时,电流越大则反芳香性越强;如果没有产生明显净电流,则是非芳香性。
核独立化学位移(nucleus-independent chemical shift,NICS[16])是被使用得最广泛的衡量芳香性的指标,其含义为设定的不在原子核位置上的磁屏蔽值的负值,负值越大,表明对磁场屏蔽越强,则芳香性越强。一般来说,如图1所示,设定的位置取在共轭环的几何中心,被称为NICS(0)。更进一步,在实际计算中有两种NICS值:各向同性值NICS(0)iso(磁屏蔽张量xx、yy、zz之和的平均)和磁屏蔽张量zz分量值NICS(0)zz(z轴垂直于环平面)。
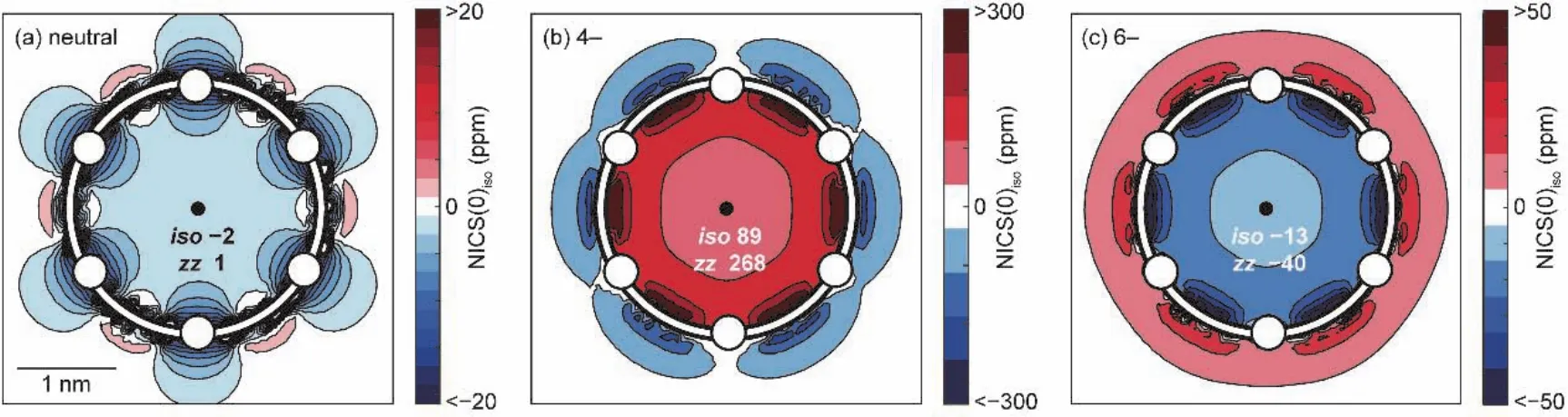
图1 卟啉纳米环NICS计算结果示意图
3 芳香性的最新进展
随着研究的深入,芳香性这一性质已经从起初的苯环衍生物,到现在体现在了不同类型的化合物上,包括卟啉类化合物、金属杂环化合物、无机金属团簇等;描述其规律的理论规则也在不断发展,逐渐趋于体系化,立体芳香性、全局芳香性等新的芳香性概念纷纷涌现。
近年来,社会互联网化进程不断加快,电子商务创新不断涌现,电子商务企业对传统商贸企业形成了激烈的跨界竞争态势。对于重庆市商贸流通业而言,顺应互联网时代要求,实现向互联网化的转变,不仅要加快构建线上销售服务能力,实现业务上网,以满足消费者日益增长的在线消费需求,更要发挥已有的传统线下优势,将线下资源与线上业务相互结合,形成线上线下互动创新发展的新兴商业模式。
3.1 立体芳香性
立体芳香性,也称三维芳香性,即三维空间上的芳香性。传统休克尔规则适用于平面芳香性,但是自C60分子被发现以来,芳香性的界限已经被拓宽到三维空间上,随着近年来莫比乌斯体系化合物合成技术的进步,立体空间上的芳香性逐渐频繁地出现在人们的视线之中。
1858年莫比乌斯等人提出莫比乌斯环的概念,虽然自然界没有莫比乌斯型分子,但是化学家一直试图去合成这类分子,其难度可想而知。2009年Kim课题组[17]总结了拓展型卟啉类化合物中的莫比乌斯芳香性和反芳香性,如果实现莫比乌斯环状芳香性,需要构象柔性好、翻转能力强、氧化还原活性高和具有潜在配位点等优势。如图2所示,在具有28个π电子的拓展卟啉结构中呈现出芳香性,这与休克尔规则恰好相反。

图2 拓展型卟啉类化合物中莫比乌斯芳香性和反芳香性[17]
既然莫比乌斯环的芳香性与平面芳香性不同,那么其他的立体结构芳香性又将如何?这一点引起了科学家持续不断的探索,而不仅拘泥于莫比乌斯型结构。2017年Kim团队[6]报道了并三噻吩桥联的共轭大环化合物1a和2a (图3),两者的区别在于1a的大环结构拓展单元为双噻吩,而2a中为并三噻吩,在同一个框架内有多个电子回路并且π电子可被不同的电路共同占有,是一种双环共轭体系。在中性状态中,形成了34个π电子和26个π电子两个相互竞争的芳环体系;在失去两个电子的氧化态中,[4n+ 1]/[4n+ 1]双自由基三重态显示出全局芳香性,即33个π电子和25个π电子的芳环体系,除去共用电子,总计40个π电子恰好符合贝尔德规则。
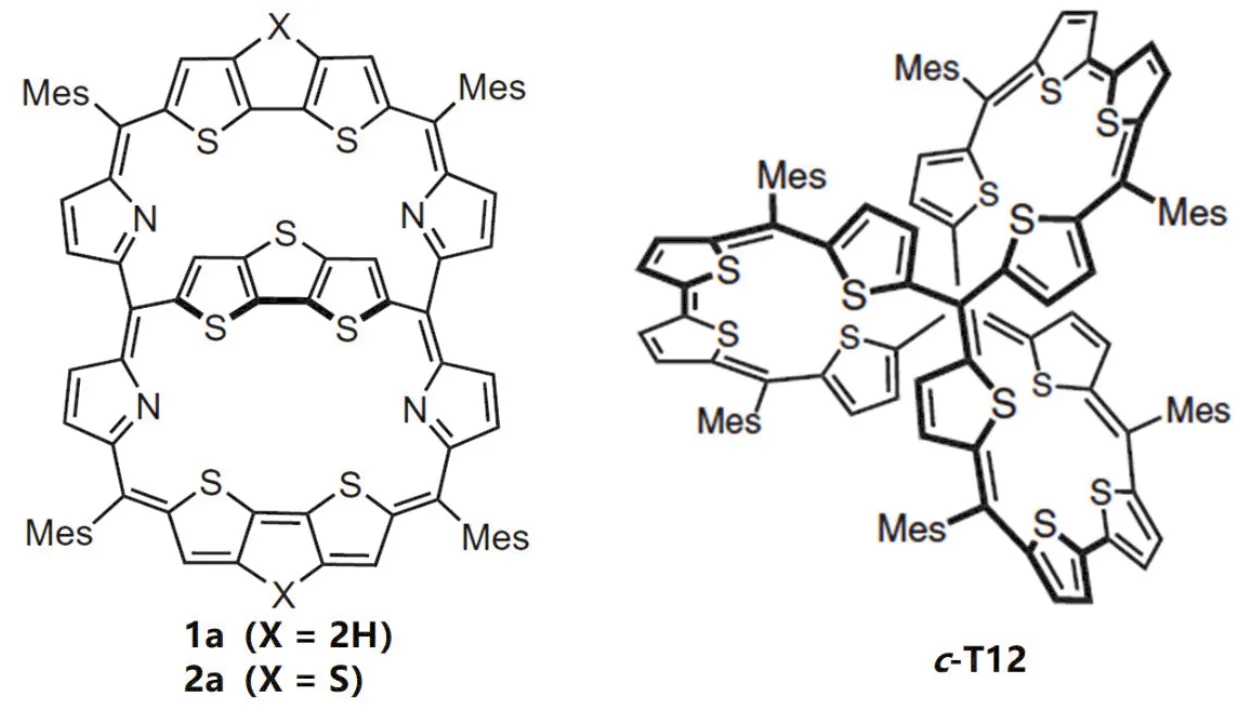
图3 噻吩类共轭大环化合物的立体芳香性[6,10]
2020年吴继善课题组[10]非常巧妙地通过Friedel-Crafts烷基化和Yamamoto偶联反应合成了一种含有12个噻吩结构的完全共轭的双自由基分子笼c-T12,可根据对称性、π电子数目和自旋态获得不同类型的芳香性。在[38π]中性状态下为单环共轭休克尔芳香性;在+2氧化态下为单环共轭贝尔德芳香性;在+4氧化态下为多环共轭立体全局反芳香性;在+6氧化态下为多环共轭立体全局芳香性。
如图3所示,两种多环共轭分子通过改变自身价态,不仅可改变芳香性的程度,而且可改变芳香性的种类,特别是分子c-T12,通过改变分子价态来改变甲基上氢质子在低场与高场中的指向,实现分子笼立体反芳香性与芳香性之间的转换,这些工作是芳香性概念发展中的里程碑。
3.2 全局芳香性
全局芳香性与立体芳香性既有联系又有区别,立体芳香性是针对平面芳香性而言,全局芳香性的概念是建立在环电流磁学判据的基础上提出的,针对局部芳香性,因而其研究对象多为具有多个基本单元的大环化合物,其中最典型是苯环类大环多自由基化合物和卟啉纳米环。
3.2.1 苯环类大环多自由基化合物中的全局芳香性
迄今为止,卟啉类化合物中已经出现了很多大环(反)芳香族化合物,但是由于苯环的π电子存在较大的共振能量,使得苯环类大环化合物无法表现出全局芳香性,因此具有全局芳香性的苯环类多环芳烃很少被合成。值得注意的是,当分子处于开壳双自由基或多自由基的形式时,它们可以适应自身的几何形状和电子自旋状态以达到最低能态,趋于芳香化。
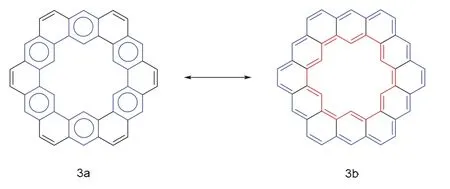
图4 凯库勒烯共振杂化体3a和3b [18]
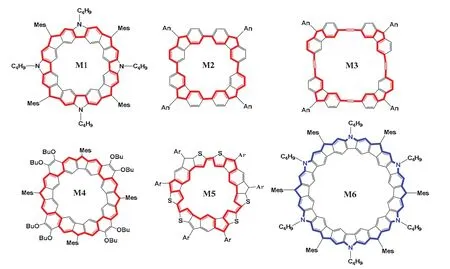
图5 系列大环多自由基化合物[19]
这些大环多自由基分子有诸多共同点,它们普遍存在两种或两种以上共振结构,在共振中存在着芳香六元环的形成与破裂,并且这些分子处于一种局部芳香性与全局(反)芳香性的平衡状态,但全局(反)芳香性往往占主导作用,可以用休克尔规则去解释他们的芳香性,却又不完全受限于休克尔规则。并且几乎所有的这种类型的大环结构都能够通过在环戊环的碳位点上添加额外的质子巧妙地实现全局芳香性与局部芳香性之间的转换,由此引发我们对局部芳香性与全局芳香性之间的转化问题的探讨。
最值得一提的是,吴继善课题组[7]合成了一类五元环和六元环交替稠合的大环多自由基化合物——具有碗状骨架8MC和近乎平面骨架10MC,在这种结构中内环与外环的行为相互独立(图6)。在碗状分子8MC中,内环有24个π电子,外环有32个π电子,处于基态的外环与内环皆有[4n]个π电子,然而根据1H NMR显示外环氢质子位于低场(δ= 11.37),内环氢质子位于高场(δ= −12.08):表明该化合物具有很强的芳香性,这与休克尔规则恰恰相反。对于10MC来说,内环和外环分别具有30和40个π电子,内环为单重态满足休克尔规则,而外环为三重态遵循贝尔德规则,对于整个大环来说具有全局芳香性的特征。对于8MC的不寻常现象,作者认为在8MC中内环与外环都具有三重态双自由基的特征,而两个三重态环耦合为一个单重态大环即“内三重态-外三重态”是这种全局芳香性形成的原因。这些研究为轮烯套轮烯结构的设计提供了合理借鉴。
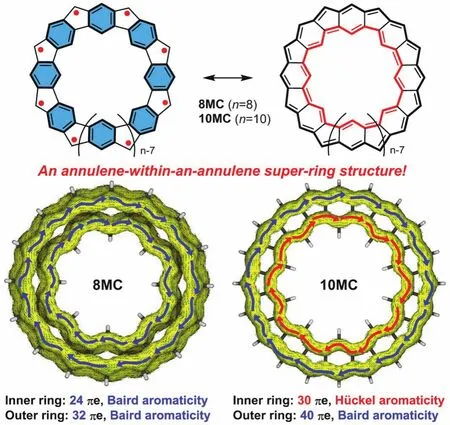
图6 具有轮烯套轮烯结构和全局芳香性的大环多自由基[7]
3.2.2 卟啉纳米环在全局芳香性中的研究
卟啉是一具有代表性的物质,据研究表明,它的π电子可以多达162个,周长可以达到16 nm[20],属于超分子环,在大环芳香性的研究中是不可或缺的,尤其是对研究大环芳香族化合物芳香性转换具有重要意义。卟啉纳米环具有优异的性质,它可以通过改变氧化态、还原态和基本组成单元的数目实现全局芳香性和全局反芳香性的转换,这对预测激发态的行为具有重要意义。
如图7所示,Martin D. Peeks和Michael Jirasek等人报道了六卟啉纳米环,分别为c-P6[20]和c-P6·T6[9]。通过改变部分卟啉基本单元氧化态来改变整体大环的氧化态从而实现改变芳香性的目的。由于中性状态下每个卟啉分子都能维持独立的环电流,因此卟啉类大环在中性时仅表现出局部芳香性,并不表现出全局(反)芳香性。对于c-P6·T6分子,当其失去4个电子时(即分子呈现+4价,80个π电子)具有全局反芳香性,当其失去6个电子时(即分子呈现+6价,78个π电子)具有全局芳香性。随后通过增加卟啉环的电子个数的方式来改变它的芳香性,发现卟啉环的两种还原态−4价(88个π电子)和−6价(90个π电子)与氧化态相似,分别表现出全局反芳香性和全局芳香性,符合休克尔规则。
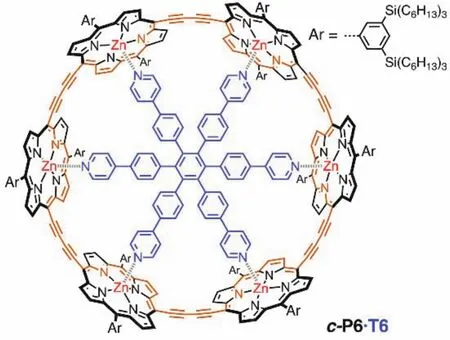
图7 卟啉纳米环中的全局芳香性和反芳香性[9]
Harry L. Anderson教授团队成功合成了具有162个π电子的卟啉纳米环,这是史上最大的芳香环[11](图8)。通过与不同的模板分子结合可以改变环的几何结构。NMR结果显示:当环几何结构为扭曲8字形时无全局环电流,这表明通过改变分子的几何结构,芳香性可以被巧妙地打开或关闭。这一结论为可芳香性分子开关的研究提出了新的解决方案。
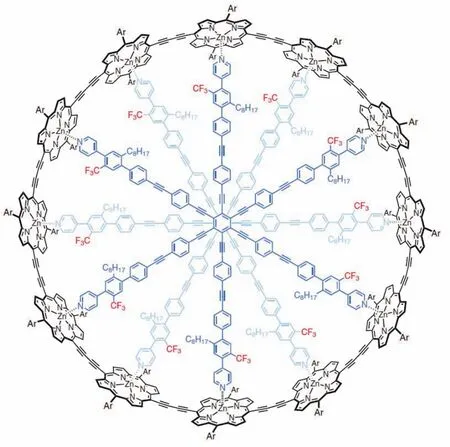
图8 目前最大芳香纳米环c-P12[b12]·(T6ef)2 [11]
除氧化还原和几何结构,电子自旋态的改变对芳香性亦有影响。激发态卟啉纳米环(反)芳香性较难被实验证明,因为NMR谱图并不适用于激发态,而大环的NICS值也因尺寸和不对称性受限。将紫外-可见吸收光谱和红外光谱用于激发态下大环化合物芳香性的评估:(1) 在用紫外吸收光谱检测时,反芳香性卟啉的三重激发态的光谱峰型显得宽而离散,不如芳香性卟啉的尖锐而有特征,类似芳香化合物基态的吸收光谱形状,但最近实验表明反芳香性卟啉也能展现尖锐的峰型,因研究过少无法证明其正确性;(2) 在用红外光谱检测卟啉单重态激发态的芳香性时,更少的红外活性振动对芳香性分子的对称做出了很好的解释;(3) 大环环电流的大小可以在某种程度上被拓扑结构所控制的,这在小环里是根本无法体现的。
总之,含有5–9个卟啉单元的纳米环含有70–126个π电子,已经超出经典的贝尔德规则范畴,但其第一激发三重态的芳香性经过密度泛函理论DFT计算证明,仍然符合贝尔德规则。例如162个π电子的大环,这些超分子环的结构仍然符合休克尔规则。与此同时,也存在诸多新问题:(1) 当纳米环在还原态和氧化态同时具有全局芳香性时,处于还原态的纳米环的全局芳香性是否一定比处于氧化态的纳米环的全局芳香性表现得更为明显?(2) 通过失去电子来实现全局芳香性的化合物是否也可以通过得到电子来达到这一效果?(3) 纳米环局部芳香性和全局芳香性切换的分子开关机制是什么?这些科学问题,都有待进一步理解芳香性的概念及芳香性强弱的影响因素。
3.3 其他芳香性
3.3.1 超共轭芳香性
1999年,理论化学家Schleyer和Nyulászi[21]通过计算化学发现,在环戊二烯中sp3杂化碳原子上两个给电子取代基可以通过超共轭作用参与环内的π共轭,形成超共轭芳香性。2016年,清华大学赵亮团队与厦门大学朱军团队合作首次报道了以过渡金属作为取代基的吲哚四核金簇中存在超共轭芳香性[5],同时他们发现过渡金属取代基比传统主族元素作为给电子取代基可以产生更为显著的超共轭芳香性效应。
3.3.2 自适应芳香性
根据贝尔德法则,一般情况下芳香性分子在第一激发三重态下会出现与基态相反的芳香性,2018年朱军等人合成了一种有机金属化合物[22](图9),此分子在S0和T1状态下同时具有芳香性。根据NICS计算、前线轨道分析和电子自旋密度的芳香性分析都得出了相同的结论:该分子在中性时与休克尔和贝尔德法则相吻合,即S0态下具有芳香性,T1态下具有反芳香性,而其阳离子状态时却得出与以往规律不同的现象:在S0和T1态下都具有芳香性,将之称为自适应芳香性。研究认为平面内的激发比平面外的π–π*作用对形成自适应芳香性更有利,此研究将过渡金属用于芳香性分子的研究中,为之后的自适应芳香性的研究和其他芳香性分子的合成提供了合理的借鉴。
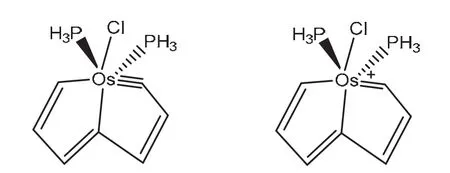
图9 锇杂戊搭烯S0和T1态的自适应芳香性[21]
4 芳香性的未来应用
由于芳香体系有独特的稳定性,众多涉及芳构化的反应拥有一些特殊的芳香驱动力[14],在光电材料领域有长足的应用。例如激发态芳香性的理论信息仍处于匮乏状态,但可用于解释光化学反应的合理性,解释液晶中的光诱导结构变化[23]。在光学材料方面,三维球形芳香性的发展也带来了不一样的思路,提出了混合共轭π共轭桥的概念。大环芳香性化合物有着很多优异的性能,例如大环多自由基化合物的小带隙可以很好地稳定多个电荷。由于离域电子波函数之间相互干涉,因此更大的芳香环可能会产生不寻常的量子效应。巨大芳香环为制造超导电流回路提供新方法,有望成为微纳环超导材料,在量子计算机的组成上有潜在的应用价值。
5 结语
本文综述了芳香性概念的最新进展,从研究背景到理论与实验的判据,特别是近五年来,大环化合物全局芳香性的研究,讨论了不同氧化态、不同激发态、不同几何结构对芳香性强弱、全局芳香性与局部芳香性之间的转换以及全局(反)芳香性之间的切换的影响,见证了休克尔和贝尔德规则对各种超出预期范围的大环化合物强大的理论包容性,同时看到了芳香性这一概念的历久弥新。
猜你喜欢
数学物理学报(2022年4期)2022-08-22
数学物理学报(2022年2期)2022-04-26
数学物理学报(2022年1期)2022-03-16
数学物理学报(2021年6期)2021-12-21
河北理科教学研究(2020年1期)2020-07-24
应用数学(2020年2期)2020-06-24
山东化工(2020年5期)2020-04-07
金桥(2018年4期)2018-09-26
中国卫生(2014年5期)2014-11-10
化工管理(2014年23期)2014-08-15
