
CLCNKB基因复合杂合新突变致成人经典型Bartter综合征一例
2021-10-13 07:32冯昌梅张海滨陈燕妮陈晓毓
中国临床新医学 2021年9期
冯昌梅, 张海滨, 陈燕妮, 陈晓毓
1 病例介绍
患者,女,31岁,因“反复乏力1个月余”入院。患者于入院前1个月余无明显诱因出现全身乏力,活动时较重,休息后可缓解,到当地医院就诊,查电解质示血钾2.6 mmol/L,予补钾处理,乏力症状缓解,但病情反复,多次外院复查血钾波动于2.6~2.8 mmol/L。为进一步诊治低钾血症,收住我院内分泌科。患者既往体健,生长发育正常,已婚,已育。无呕吐、腹泻、利尿剂及化疗药物使用史,无酗酒史,无类似疾病家族史。父母体健,非近亲结婚。体格检查:身高168 cm,体重50 kg;体质量指数(Body Mass Index,BMI)17.72 kg/m2;血压96/71 mmHg。智力发育正常,高中学历。双眼无突出,双侧甲状腺无肿大,心肺腹部查体未见异常,四肢肌力五级,肌张力正常,生理反射存在,未引出病理反射。在院期间血压监测(住院9 d,血压监测17次):最高123/79 mmHg,最低90/69 mmHg,平均血压(102.06±9.51)/(72.65±9.70)mmHg。辅助检查:24 h血电解质:钾2.6~2.9 mmol/L,镁0.88~0.95 mmol/L,钙2.15~2.50 mmol/L,钠136~139 mmol/L,氯95~96 mmol/L,总二氧化碳32.4~34.8 mmol/L。24 h尿电解质:钾14.89~23.13 mmol/L,钙1.23~1.61 mmol/L,钠99.7~138.8 mmol/L,氯75.1~112.2 mmol/L,尿磷3.9~5.3 mmol/L。24 h尿游离皮质醇测定:91.28~135.1 μg/24 h。血气分析:pH值7.454,实际碳酸氢根28.5 mmol/L,标准碳酸氢根28.2 mmol/L,剩余碱4.5 mmol/L。甲功6项、肝功能正常,肌酸激酶及同工酶正常。血皮质醇在8:00、16:00、24:00分别为14.22 μg/dl、2.51 μg/dl、1.95 μg/dl。血促肾上腺皮质激素(adrenocorticotropic hormone,ACTH)在8:00、16:00分别为35.52 pg/ml、36.98 pg/ml。肾素活性卧位为21.497 ng/(ml·h)[0.15~2.33 ng/(ml·h)],立位为20.19 ng/(ml·h)[0.10~6.56 ng/(ml·h)]。醛固酮卧位为0.209 ng/ml(0.03~0.16 ng/ml),立位为0.282 ng/ml(0.07~0.30 ng/ml)。血尿粪常规、皮质醇、ACTH、肝功能正常,肌酸激酶及同工酶正常。常规心电图、胸部正侧位片、泌尿系超声、肾上腺彩超未见异常。考虑患者低钾血症合并代谢性碱中毒、尿钾增多、醛固酮及肾素均较高,但血压未见明显异常,故考虑“Bartter综合征(Bartter′s syndrome,BS)”或“Gitelman综合征”可能性大,与患者及家属充分沟通告知病情并签署知情同意书后,留取患者及其父母外周静脉血送福州金域医学检验所行高通量测序。应用过柱法从检样中提取DNA,对BSND、CLCNKA、CLCNKB、KCNJ1、SLC12A1、SLC12A3、CASR等基因的外显子区(编码区)序列进行直接测序,与参考序列进行比较,从而发现可能存在的基因突变。结果显示患者携带CLCNKB基因一个杂合致病变异和一个临床意义未明的杂合变异,分别遗传自父母。其父亲、母亲分别在CLCNKB基因检测到c.877G>A杂合变异、c.1093delC杂合变异(见图1)。根据美国医学遗传学与基因组学学会联合美国分子病理学会2015年制订的“基因序列变异的解释标准和指南”进行致病性分析[1]。(1)c.1093delC突变为移码突变,突变会导致所编码的蛋白质发生截短从而丧失其正常功能(非常强的致病性证据,PVS 1)。(2)c.1093delC在HGMD数据库、ESP6500siv2_ALL、千人基因组(1000g2015aug_ALL)和dbSNP147数据库均未见收录(中等致病性证据,PM 2)。(3)c.1093delC突变来源于患者母亲,同时在另一条染色体上发现了CLCNKB基因存在1个已知错义突变(c.877G>A),来源于其父亲,符合隐性遗传病的遗传规律(中等致病性证据,PM 3)。(4)患者的临床表现与CLCNKB基因突变导致的经典型BS表型高度吻合(支持性致病性证据,PP 4)。c.1093delC突变的证据强度为“PVS 1+PM 2+PM 3+PP 4”,属于致病性突变。同理,c.877G>A突变为错义突变,突变来源于患者父亲,虽然dbSNP147数据库有收录,但在正常人群中的突变频率极低(0.00008,rs748680754),且生物信息学工具程序PolyPhen2和SIFT预测其致病可能性较大,故证据强度也为致病性突变。以“经典型Bartter综合征CLCNKB基因”检索万方、中国知网、重庆维普数据库,“Bartter′s syndrome CLCNKB”检索PubMed数据库文献,并未发现有关c.1093delC和c.877G>A突变位点的文献报道。通过以上检索证实c.1093delC+c.877G>A为一种新的CLCNKB基因复合杂合突变。治疗上予氯化钾缓释片1.0 g口服,3次/d,螺内酯20 mg口服1次/d。10余天后(2020年1月6日),患者乏力症状明显改善,复查血钾3.2 mmol/L,血镁、钠、钙、氯、总二氧化碳基本正常。2个月后(2020年3月7日)复诊,无乏力不适,复查血钾3.2 mmol/L,4月余后(2020年5月9日)复诊无乏力,血钾2.9 mmol/L。
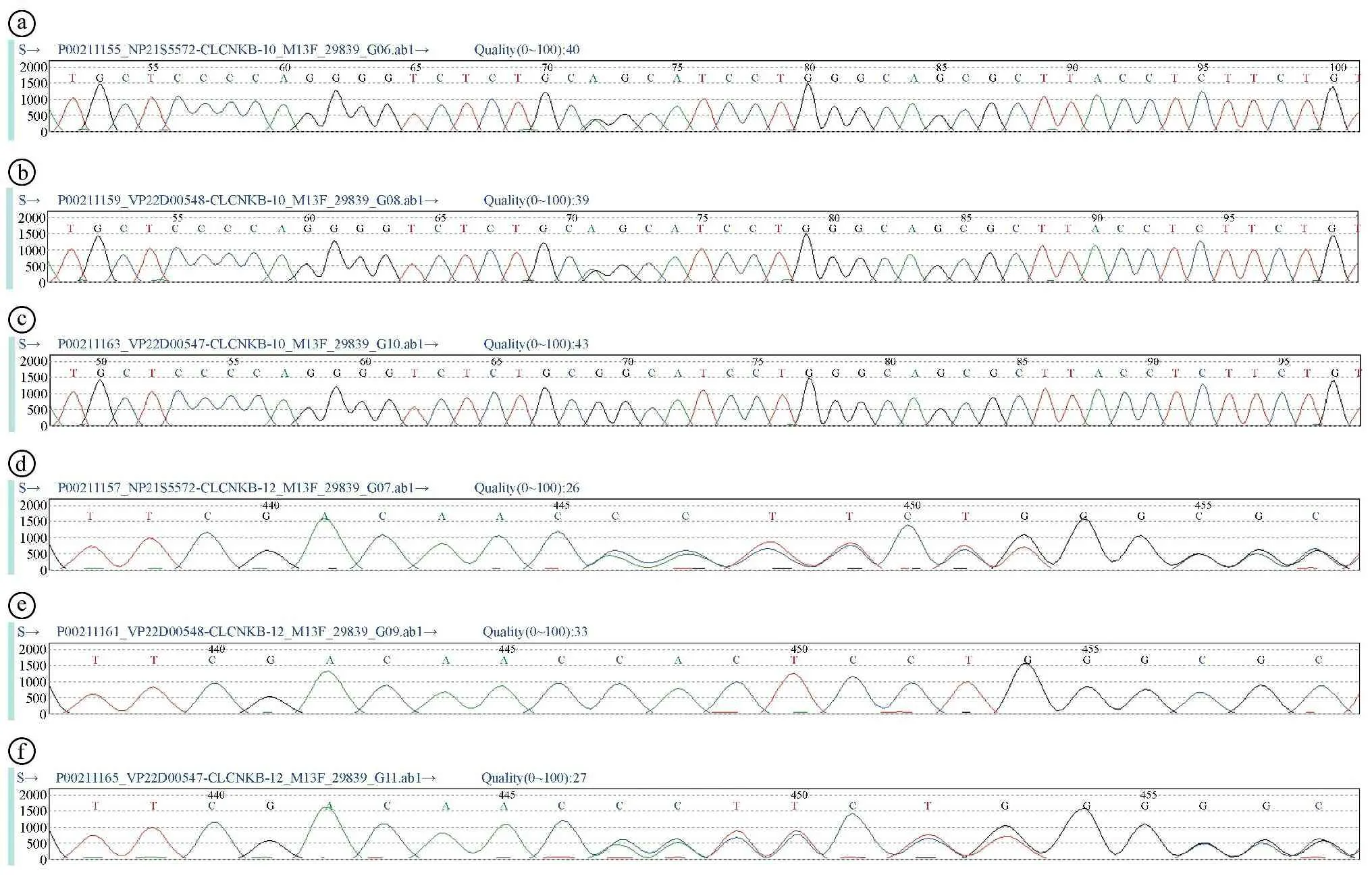
ⓐ患者测序图CLCNKB Exon10 c.877G>A(p.Gly293Ser)(检出);ⓑ患者父亲测序图CLCNKB Exon10 c.877G>A(p.Gly293Ser)(检出);ⓒ患者母亲测序图CLCNKB Exon10无突变;ⓓ患者测序图1p36.13 Exon12 c.1093delC(p.His365fs)(检出);ⓔ患者父亲测序图1p36.13 Exon12无突变;ⓕ患者母亲测序图1p36.13 Exon12 c.1093delC(p.His365fs)(检出)
2 讨论
2.2BS目前无法根治,主要治疗手段以对症治疗为主,包括纠正电解质紊乱、抑制肾素水平升高及高醛固酮血症。补钾建议使用氯化钾,钾盐(如柠檬酸盐)可能会加重碱中毒。血钾的目标水平尚不明确,合理的目标水平可能是3.0 mmol/L[2],经典型BS的低钾血症比Ⅰ型、Ⅱ型BS更难纠正[4,7,15]。非甾体类抗炎药如吲哚美辛等,可抑制肾素、醛固酮的活性,提高血钾水平。虽然一项纳入42例中国经典型BS患者的研究中近50%患者必须使用螺内酯才能达到令人满意的血钾水平[4],但事实上盐皮质受体拮抗剂是否有助于维持血钾水平目前仍存在争议[17]。大多数生长激素缺乏的报道都与经典型BS相关,但一份报告表明BS患者在开始使用COX抑制剂之前,重组人生长激素补充对生长激素缺乏症没有反应[4]。本例患者生长发育正常,明确诊断前予口服氯化钾处理,血钾水平波动于2.6~2.9 mmol/L,明确诊断后予口服氯化钾及螺内酯血钾水平波动于2.9~3.2 mmol/L。
综上所述,BS患者通过基因检测发现CLCNKB基因新型c.1093delC+c.877G>A突变,丰富了经典型BS突变谱,今后需进一步完善功能试验以明确该突变与疾病的内在联系。
猜你喜欢
种子(2021年3期)2021-04-12
透析与人工器官(2020年1期)2020-11-16
中华养生保健(2020年2期)2020-11-16
中国循环杂志(2020年4期)2020-04-27
养生大世界(2019年12期)2019-12-11
心肺血管病杂志(2019年12期)2019-05-20
家庭科学·新健康(2018年8期)2018-10-30
外语教学理论与实践(2016年1期)2016-06-11
现代检验医学杂志(2015年5期)2015-02-06
河南医学研究(2014年4期)2014-02-27
