
超低负载量Au/g-C3N4@CeO2室温催化去除甲醛性能*
2021-10-14 01:04严朝雄徐志花
功能材料 2021年9期
李 琴,严朝雄,黄 刚,石 零,徐志花
(江汉大学 工业烟尘污染控制湖北省重点实验室,武汉 430056)
0 引 言
甲醛(HCHO)作为典型的室内空气污染物,不仅会刺激眼睛和喉咙,还会导致人体皮肤瘙痒等疾病,如果长期接触,甚至会致癌及死亡。2004年1月,甲醛被国际癌症研究机构(IARC)列为人类致癌物(Ⅰ类)[1]。有效治理室内空气中的甲醛污染,对保障人体健康具有重要意义。
目前,室温去除甲醛技术因其无需提供额外的能量和装置而能将甲醛彻底氧化为二氧化碳和水,引起了研究工作者的兴趣。高效催化剂的设计和制备是室温去除甲醛的关键,其类型主要有负载贵金属型或过渡金属氧化物两大类[2]。TiO2、MnOx、Co3Ox、CeOx等过渡金属氧化物对甲醛的催化降解表现出较好的活性[3-5],但操作温度较高或降解所需时间较长。负载贵金属型催化剂主要是将Pt、Au、Pd和Ag等贵金属负载在TiO2、Fe2O3、MnO2、NiO和CeO2等过渡金属氧化物上[6-15],该类催化剂具有活性高、稳定性好及操作温度低等特点,是当前研究的热点。众多的研究成果表明,载体微结构及表面羟基含量等性质对负载贵金属型催化剂的催化性能具有重要影响。近年来,人们在不影响复合催化剂催化性能的基础上,致力于优化载体的微观结构和组分,降低贵金属含量,以达到商业化应用。例如,在Pt/TiO2催化剂中Pt含量由1%(质量分数)降低至0.1%(质量分数)仍能实现近100%的甲醛转化率[16-17]。人们发现除了Pt外,贵金属Au也被开发作为负载型催化剂用于甲醛的去除中。例如,Chen等人制备的Au/CeO2(Au含量为1%质量分数)可以在室温下达到接近100%的甲醛转化率[18]。Yan等人发现Au/AlOOH(Au含量为0.8%(质量分数))能在室温下高效去除甲醛[19]。然而,贵金属Au含量相较于实际应用仍然较高,开发高效、价格低廉和环境友好的载体进而降低贵金属Au使用量具有十分重要的研究意义。
类石墨烯氮化碳(g-C3N4)是一种非金属半导体光催化剂,具有和石墨烯相似的层状结构,其表面大量共轭双键有利于贵金属Au纳米颗粒的稳定和高度分散。此外,其原料经济易得,对环境友好。CeO2具有很强的储存和释放氧能力,氧通过在不同金属氧化态之间快速切换,从而促进氧化反应的发生。近期的研究工作表明,Au在甲醛氧化中的活性与CeO2结合后显著增强[20]。鉴于上述优点,本课题通过煅烧法,改变三聚氰胺使用量制备得到g-C3N4@CeO2载体,然后通过浸渍法负载超低Au含量(0.003%(质量分数))的Au/g-C3N4@CeO2催化剂,并研究了催化剂在室温下催化氧化甲醛的性能。
1 实验部分
1.1 催化剂的制备
Au/g-C3N4@CeO2的制备:取2.5 g尿素,2.0 g六水合硝酸铈,于烧杯中混合搅拌成糊状后分别加入三聚氰胺(0 、2.0、3.0 、4.0 g)继续搅拌均匀,混合物转移至坩埚于马弗炉中550 ℃下煅烧2 h(升温速率为2 ℃/min)。分别取上述0.4 g煅烧后的产品,分散于30 mL蒸馏水中,在磁力搅拌器上搅拌均匀后加入0.25 mL 0.1 g/L氯金酸(即Au理论含量为0.003%(质量分数)),搅拌30 min后加入5 mL 0.1 mol/L的NaOH/NaBH4混合溶液,继续充分搅拌30 min,随后离心并用去离子水洗涤3次,离心产物置电热鼓风干燥箱中在70 ℃下干燥12 h,所得干燥产物充分研磨后依次标记为Au1、Au2、Au3、Au4。
Au/g-C3N4的制备:取3.0 g三聚氰胺固体于坩埚,放置于马弗炉中550 ℃下煅烧2 h(升温速率为2 ℃/min),取0.4 g煅烧后的产品,分散于30 mL蒸馏水中,在磁力搅拌器上搅拌均匀后加入0.25 mL 0.1 g/L氯金酸(即Au含量为0.003%(质量分数)),搅拌30 min后加入5 mL 0.1 mol/L的NaOH、NaBH4混合溶液,继续充分搅拌30 min,随后离心并用去离子水洗涤3次,离心产物置电热鼓风干燥箱中在70 ℃下干燥12 h,所得干燥产物充分研磨后标记为Au5。
g-C3N4@CeO2的制备:取2.5 g尿素,2.0 g六水合硝酸铈,于烧杯中混合搅拌成糊状后加入三聚氰胺3.0 g继续搅拌均匀,混合物转移至坩埚于马弗炉中550 ℃下煅烧2 h(升温速率为2 ℃/min),把煅烧后的产品标记为S1。所制备催化剂的详细情况如表1所示。

表1 制备催化剂所使用的原料情况Table 1 The preparation precusors of catalyst
1.2 催化剂的表征
通过X射线衍射(XRD)、比表面积测试(BET)、透射电镜(TEM)、红外光谱(FTIR)、拉曼光谱(Raman)等手段对催化剂的理化性能进行表征和分析。采用红外光声光谱气体检测仪(INNOVA Air Tech Instruments Model 1412)测试催化剂室温催化氧化甲醛的性能。
1.3 催化剂性能测试
1.3.1 催化剂性能测试
通过红外光声光谱气体检测仪实时监测反应箱中HCHO、CO2、CO和H2O等气体浓度随测试时间增加而发生的变化。室温下,将0.1 g催化剂均匀分散在直径为14 cm的玻璃培养皿中,用玻璃片盖住培养皿后放入红外光声光谱气体检测仪的反应箱中,然后密封反应箱并向反应箱中注入适量甲醛溶液。用5 W的电风扇加快甲醛气体的释放和流动,待反应箱内甲醛气体的浓度稳定后,移去培养皿上的玻璃片,开始实验。
所制备催化剂对甲醛的催化性能由甲醛去除率η表示,用式(1)计算:
(1)
其中,[HCHO]s代表甲醛的初始浓度,[HCHO]f代表甲醛的终止浓度。
1.3.2 催化剂重复性能测试
室温下,将样品Au1按催化剂性能测试的条件进行重复测试。测试完毕后将样品密封保存于干燥箱中,每次测试前将样品置于电热鼓风干燥箱中于80 ℃下干燥1 h进行催化剂活性的再生。如此循环,共测试5次。
2 结果与讨论
2.1 XRD谱图分析
催化剂的XRD谱图如图1所示。图中样品Au1、Au2、Au3、Au4和S1的XRD谱线峰位相似。在2θ等于28.6°、33.1°、47.6°、56.3°、68.9°和76.5°处的衍射峰分别对应于CeO2的(111)、(200)、(220)、(311)、(400)和(331)晶面(JCPDS 34-0394)[21],显示了CeO2的立方萤石结构。其中,样品Au3和S1的谱线高度一致,且前者XRD图谱中未观察到Au的衍射峰(最强衍射峰位于38.1°,JCPDS 04-0784),这与催化剂中Au的负载量比较低和Au纳米颗粒具有较小的尺寸并高度分散在样品表面有关。Au5样品谱线中,在2θ=27.2°处的衍射峰对应于石墨相氮化碳的(002)晶面(JCPDS 87-1526),该处的XRD衍射峰归属于氮化碳共轭芳香体系的层间堆积峰[22],证明本文制备方法中三聚氰胺主要生成g-C3N4。在Au1、Au2、Au3、Au4和S1的XRD谱图中,未观察到明显的g-C3N4的衍射峰,这或许是因为g-C3N4(002)的衍射峰与CeO2(111)的衍射峰部分重叠或氮化碳含量较低有关。

图1 样品Au1、Au2、Au3、Au4、Au5、S1的XRD谱图Fig 1 XRD patterns of Au1,Au2,Au3,Au4,Au5 and S1 of the samples
2.2 拉曼图谱分析
为进一步了解催化剂分子内部结构信息和振动模式,对样品Au3、Au5、S1 进行拉曼图谱分析。如图2所示,样品Au3、S1在465 cm-1处均有一个尖锐的拉曼峰,对应于空间群为Fm3m立方萤石结构的Ce-O-Ce键对称伸缩振动的F2g拉曼峰[23]。相较于样品S1,样品Au3的拉曼峰有显著增强,原因是贵金属Au纳米结构的局域表面等离子体共振效应增强了拉曼散射光谱,由此证明Au已成功附着在样品Au3上。

图2 样品Au3、Au5、S1的Raman谱图Fig 2 Raman spectra of Au3,Au5 and S1 of the samples
2.3 TEM图分析
如图3(a)所示,样品Au3由一些无规则形状的薄片组成,其表面存在大量细小纳米颗粒。催化剂中存在较大空隙,这些空隙可以为反应气体分子、中间产物和生成物的交换提供便捷的路径,有利于提高甲醛的催化转化。从图3(b)中可以观察到分散在g-C3N4薄片上的大量约50~100 nm左右的CeO2颗粒。因为Au的含量较少,较难观察到Au的纳米颗粒。

图3 样品Au3的TEM全貌图(a)及放大图(b)Fig 3 TEM image and enlarged TEM image of sample Au3
2.4 红外光谱分析
催化剂的红外光谱如图4所示,在所有样品的红外光谱曲线中,位于3440 cm-1处和1636 cm-1附近的红外吸收峰分别是吸附在样品表面的水分子的O-H伸缩振动和弯曲振动而引起的。样品Au2、Au3、Au4、Au5和S1在3 000~3 300 cm-1范围内的红外宽频带峰是由于未缩合胺基的N-H波段伸缩振动,1200~1600 cm-1范围内的红外光谱峰是g-C3N4碳氮杂环伸缩振动引起的,在810和890 cm-1处出现的红外振动吸收峰归属于g-C3N4三嗪结构的红外特征峰[24]。这些现象表明了在样品Au2、Au3、Au4、Au5和S1中存在g-C3N4。样品Au1、Au2、Au3、Au4和S1在540 cm-1处的峰属于金属氧化物的Ce-O振动[22]。以上结果更进一步说明了Au2、Au3、Au4和S1中g-C3N4和CeO2成功复合在一起。通过对比S1,Au3图在540 cm-1处峰发生了明显偏移,这与Au3催化剂中的成分间的强相互作用有关。

图4 样品Au1、Au2、Au3、Au4、Au5、S1的红外光谱图Fig 4 Infrared spectra of Au1,Au2,Au3,Au4,Au5 and S1 of the samples
2.5 BET分析
图5为样品Au1、Au2、Au3、Au4、Au5、S1的N2吸附-脱附等温线与相应的孔径分布曲线。图5(a)的 N2吸脱附曲线中,所有样品均存在Ⅳ型N2吸脱附等温曲线和H3型滞后环[25],表明所有制备样品具有介孔结构。图5 (b)的孔径分布图中,所有样品的孔径分布主要集中在2.0~6.0 nm附近,进一步证实介孔的存在。样品具体的物理特性测量值如表2所示,样品Au2、Au3和S1均具有较大的比表面积和孔容。一般来说,较大的比表面积和孔容体积有利于贵金属纳米粒子的分散,同时可为HCHO气体分子提供更多的吸附位点或反应活性位点,从而提高催化剂对甲醛的催化活性。通过比较不同样品的甲醛催化降解效率,我们发现样品Au2、Au3具有相对较大的比表面积(分别为72和67 m2/g)和孔容(分别为0.12和0.13 cm3/g)而有更高的降解效率。由于Au的密度大于其载体密度,因此负载Au样品比其载体材料的密度更大,导致这些样品(Au1、Au2、Au3、Au4、Au5)的比表面积与孔容均有所下降。此外,在负载Au 的过程中,Au 纳米颗粒以及原料(NaOH、NaBH4)中的Na+会覆盖或部分堵塞载体的孔道,也会导致载Au样品比表面积下降[26]。S1样品虽然具有最大的比表面积和孔容但由于未负载贵金属Au纳米粒子,无法为甲醛的反应提供活性位点,所以对甲醛的催化降解效率最低。
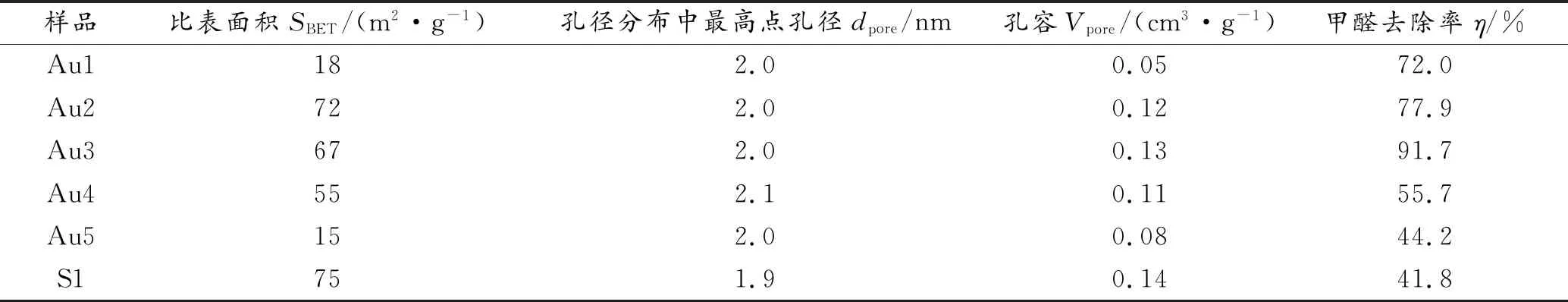
表2 样品的比表面积、孔径、孔容及甲醛去除效率Table 2 Specific surface area,pore size,pore volume and formaldehyde removal efficiency of the samples

图5 样品Au1、Au2、Au3、Au4、Au5、S1的N2吸附-脱附等温线(a)与相应的孔径分布曲线(b)Fig 5 N2 adsorption-desorption isotherm and pore size distribution curve of samples Au1,Au2,Au3,Au4,Au5 and S1
2.6 XPS分析
利用XPS对样品的表面元素组成和表面化学态进行了分析。图6(a)给出了样品Au1、Au3和S1的XPS全谱。从图中可以看出样品中均有Ce,O,C与N元素,对应结合能分别为882.3、916.7(Ce3d)、529(O1s)、284.8(C1s)与398.4 eV(N1s)。仔细观察发现,Au1和Au3在84 eV的结合能位置处出现了很小的Au4f光电子峰,表明样品含有Au元素,进一步表明样品被成功制备。图6(b)给出了样品Au1、Au3和S1中Ce3d的高分辨XPS图谱,经过拟合,在该范围内可以拟合出10个峰:结合能位于880.6,884.4,899.3和903.1 eV处的峰归属于Ce3+;结合能位于888.2,888.6,898,900.7,907.2,916.1 eV处的峰归属于Ce4+,表明催化剂中同时存在Ce3+和Ce4+[27-29]。由图中Ce3+峰面积大小可以看出,样品Au3表面的Ce3+含量高于Au1和S1,Ce3+含量越高,表明其氧缺陷越多,后续衍生的活性氧物种也就越多。图6(c)对O1s的高分辨XPS图谱进行了拟合,位于528.7~529 eV处的峰属于晶格氧,位于530.5~531 eV处的峰属于表面吸附氧物种[30],位于531.8~532.2 eV处的峰属于表面羟基基团。羟基氧和表面吸附氧物种的存在可以促进甲醛吸附及后续的氧化。图6(d)给出了样品Au3中Au4f的高分辨XPS图谱,Au4f7/2与Au4f5/2的结合能分别为83.0与86.6 eV,其结合能差为3.6 eV(金属态Au的特征),表明样品中Au3中存在Au元素,并且主要以金属态存在。文献中报道Au4f7/2与Au4f5/2的结合能分别为85.0与87.7,与之相比,样品Au3中Au4f的结合能发生了负移动,这是因为金属和载体发生了强烈的相互作用,在此作用下,CeO2中的电子迁移到Au上[31],这也表明Au和 CeO2之间存在的强烈相互作用,该相互作用也有利于产生更多的氧空位[32]。对于甲醛催化氧化反应来说,金属态的Au比其他氧化态的Au能提供更多的反应活性位[20]。

图6 样品Au1、Au3和S1的XPS全谱(a);样品Au1、Au3和S1中Ce3d(b)和O1s(c)的高分辨XPS图谱;样品Au3中Au4f的高分辨XPS图谱(d)Fig 6 XPS survey spectra of samples Au1,Au3 and S1(a);high-resolution XPS spectra of the Ce3d(b)and O1s(c)regions of samples Au1,Au3 and S1;high-resolution XPS spectra of the Au4f regions of sample Au3(d)
2.7 室温催化甲醛性能分析
图7为所制备催化剂在室温下催化降解甲醛的性能图。如图7(a)所示,经过180 min的催化氧化,样品Au3对HCHO表现出显著的去除效果,甲醛浓度从1.415 ×10-4下降到1.17×10-5,去除率高达91.7%。同时,图7(b)中样品Au3的ΔCO2的浓度不断上升,从0增长到2×10-4,表明催化剂对甲醛的去除主要为催化作用,即甲醛完全转化为CO2。此外,样品Au1、Au2、Au4样品也能明显去除甲醛,但去除效率均低于样品Au3,表明样品组分中适当的g-C3N4含量对催化剂的催化性能有显著的影响:g-C3N4含量太高会使催化剂载体发生团聚,含量太低则无法很好地分散CeO2和贵金属Au颗粒。随着g-C3N4含量的改变,CeO2的含量在各样品中也不完全相同,在样品Au1中,CeO2的含量最高,几乎不含有g-C3N4却也具有较好的甲醛去除率。通过BET的数据也可知,由于不含有g-C3N4,Au1的比表面积低孔容小,不利于贵金属的分散,最终的性能测试结果也表明甲醛去除效果不是最好。在样品Au5中,仅仅含有g-C3N4,不含有CeO2,在相同时间内虽然也能一定程度降低甲醛的浓度,但是相应的CO2浓度并没有明显增加,因此样品Au5对甲醛的去除可能因为氧化产物为CO或甲酸盐,或催化剂本身具有一定的物理吸附作用。综上可得,Au/g-C3N4@CeO2催化剂对甲醛的去除效果远好于Au/g-C3N4,表明CeO2的存在对提高催化剂的活性具有积极作用,CeO2优异的储氧能力大大增强了催化剂的电荷转移,促进界面附近氧空位和Au3+的产生,促进甲酸盐的形成。甲酸盐的转化可以遵循分解和直接氧化途径,即Au3+与氧空位共存显著增强,进而提高催化剂的催化活性[20]。同时,样品Au3比S1具有更高催化活性证明Au纳米粒子的负载对提高样品的催化活性有显著的引发效果,这与Au纳米粒子提供更多的活性位点以及增强催化剂中的电荷转移有关。

图7 样品Au1、Au2、Au3、Au4、Au5、S1在催化氧化甲醛过程中随反应时间增加甲醛浓度的变化曲线(a)与ΔCO2浓度的变化曲线(b)Fig 7 In the process of catalytic oxidation of formaldehyde increases with reaction time curves of the concentration of formaldehyde and Δ changes in CO2 concentration curve of samples Au1,Au2,Au3,Au4,Au5 and S1
我们将对甲醛具有最佳催化去除效率的Au3样品按照上面相同的性能测试条件重复实验5次,每次甲醛的去除率如图8,样品Au3在五次催化去除甲醛后,对甲醛的去除率仍然保留在60.0%以上,具有较稳定的催化活性。

图8 样品Au3循环性能测试Fig 8 Cycle performance test of sample Au3
为了探寻HCHO催化氧化反应的中间产物和过程,对样品Au3进行了原位红外光谱测试,在室温下将样品置于HCHO和O2的混合气流中,如图9所示,分别在1 085、1 245、1 380、1 585、2 850和3 690 cm-1处观察到了吸收峰。位于1 085 cm-1处的吸收峰归属于二氧亚甲基(DOM)中的ν(CO)振动,位于1 245 cm-1处的吸收峰是由于DOM中的ν(CH2)与分子吸附的HCHO中的ω(CH2)重叠所致,位于1 380 cm-1处的吸收峰对应于吸附态甲酸盐物种的δ(CH)和νs(OCO)的振动,位于1 585 cm-1处的吸收峰是由于甲酸盐中的COO不对称伸缩振动引起,位于2 850 cm-1处的吸收峰是吸附的HCHO中的C-H键伸缩振动引起的[33-34]。以上结果表明甲醛在反应过程中被氧化成甲酸盐和DOM中间产物。同时,随着反应时间的增加,在3 690 cm-1处的吸收峰强度逐渐增大,表明在样品Au3表面的羟基基团逐渐增多,并参与了氧化反应。文献中也有报道羟基基团在甲醛催化氧化过程中起到了至关重要的作用[35]。甲酸盐物种能够直接地与羟基反应并被氧化,结合甲醛性能测试结果,甲酸盐和DOM最终完全分解成为CO2和H2O。

图9 样品Au3在HCHO+O2混合气中室温催化氧化甲醛的原位红外光谱Fig 9 Dynamic change of in situ DRIFTS of sample Au3 in gaseous HCHO+O2 at room temperature
3 结 论
(1)本工作所制备的Au/g-C3N4@CeO2(Au含量为0.003%(质量分数))催化剂具有价格低廉、制备工艺简单、催化活性高和稳定性较好等特点。催化剂Au3在性能测试和循环性能测试中均表现出良好的催化活性和催化稳定性,其甲醛去除率在180 min能达到91.7%。Au3相较于其他含Au样品具有相对较大的比表面积(67 m2/g)和孔容(0.13 cm3/g),有利于贵金属纳米粒子的分散而有更高的降解效率。
(2)催化氧化反应过程中甲酸盐和DOM是其中最重要的中间产物,Au与C3N4@CeO2之间由于很强的相互作用,促进界面附近活性氧物种的产生,促进甲酸盐的形成,并最终完全分解为CO2和H2O。
(3)g-C3N4能够增大催化剂的比表面积和孔容而更有利于CeO2和贵金属Au的分散。CeO2具有很强的储存和释放氧的能力,氧通过在不同金属氧化态之间快速切换,从而促进氧化反应的发生,对提高催化剂的活性具有积极作用。
猜你喜欢
分子催化(2022年1期)2022-11-02
贵金属(2021年1期)2021-07-26
贵金属(2021年1期)2021-07-26
煤气与热力(2021年2期)2021-03-19
——庆祝中国共产党成立一百周年贵金属纪念币展
中国钱币(2021年4期)2021-02-26
中国蜂业(2018年4期)2018-05-09
当代化工研究(2016年6期)2016-03-20
物理化学学报(2015年7期)2015-12-30
橡胶工业(2015年7期)2015-08-29
橡胶工业(2015年8期)2015-07-29
