
可疑植物制品中合成大麻素5F-EDMB-PICA 的鉴定
2021-12-07 03:05孟鑫花镇东贾薇刘翠梅刘杉陈月猛
法医学杂志 2021年4期
孟鑫,花镇东,贾薇,刘翠梅,刘杉,陈月猛
1.公安部禁毒情报技术中心 毒品监测管控与禁毒关键技术公安部重点实验室,北京 100193;2.贵阳市公安局禁毒支队,贵州 贵阳 550081
近年来,新精神活性物质(new psychoactive sub⁃stance,NPS)问题呈高速发展蔓延态势,新品种层出不穷,滥用形势日趋严峻[1-3]。合成大麻素是NPS中包含种类最多、更新换代最快的类别。截至2020年8月,联合国毒品和犯罪问题办公室(United Nations Office on Drugs and Crime,UNODC)统计各国发现的NPS已达1 004种,其中包含合成大麻素300余种[1-2]。截至2020 年,国家毒品实验室累计发现的NPS 达317 种,其中合成大麻素为101种,占31.9%[4]。
合成大麻素是人工合成的大麻素受体(cannabi⁃noid receptor,CB)的激动剂,从结构上可分为苯甲酰基吲哚类、苯乙酰基吲哚类、萘甲酰基吲哚类、萘甲基吲哚类、萘甲酰基吡咯类、二苯并吡喃类、苯己基酚类、吲哚酰胺类、吲唑酰胺类等。合成大麻素作用于人体中枢型受体CB1和末梢型受体CB2,效果与天然大麻素类似,但致幻效果更强,导致吸食者自残、自杀的案例时有发生。根据欧洲毒品和毒瘾监测中心(European Monitoring Centre for Drugs and Drug Ad⁃diction,EMCDDA)报告显示,2014 年,合成大麻素N-(1-甲氧基羰基-2,2-二甲基丙基)-1-(4-氟苄基)吲唑-3-甲酰胺(MDMB-FUBINACA)在俄罗斯造成了超过600 人中毒,15 人死亡;2016 年,合成大麻素N-(1-甲氧基羰基-2,2-二甲基丙基)-1-(环己基甲基)吲哚-3-甲酰胺(MDMB-CHMICA)在欧洲造成25人重度中毒,28 人死亡[5]。近年来,含有合成大麻素类物质的“小树枝”“烟叶”“烟丝”“娜塔莎”“电子烟油”等新型毒品在国内市场上不断涌现[6-10],其主要滥用方式是喷涂于烟丝、花瓣等植物表面或溶于电子烟油吸食。截至2020年3月底,新疆公安机关共破获含合成大麻素的“娜塔莎”案件上百起,相关犯罪案件急剧上升[9]。
近年来,合成大麻素制品在国内市场上不断涌现,由于其含量相对较低,新结构化合物不断出现且对应的标准品难以及时获得,给化合物的定性分析工作带来了很大的挑战[6-8]。近期,本实验室收到地方公安部门送检的1 份植物样品,经气相色谱-质谱法(gas chromatography-mass spectrometry,GC-MS)分析,推测其可能含有合成大麻素成分,但与NIST 谱库及实验室自建NPS 谱库检索比对无匹配结果。由于缺乏标准品和标准谱库,仅依靠质谱图无法确定其准确结构。对于未知化合物的结构确证,一般需要得到一定量的纯度较高的样品,并综合利用质谱、核磁共振波谱、红外光谱等进行结构解析[11-13]。为了得到高纯度未知物样品,本研究建立了采用制备液相从低纯度植物制品中分离、纯化合成大麻素的方法,并采用GC-MS、超高效液相色谱-四极杆飞行时间质谱(ultra-high performance liquid chromatography-quad⁃rupole time-of-flight mass spectrometry,UPLC-QTOFMS)、核磁共振(nuclear magnetic resonance,NMR)对制备样品进行结构解析,拟为植物制品中低含量未知合成大麻素的定性分析提供解决方案。
1 材料与方法
1.1 主要仪器与试剂
GCMS-QP2010 气相色谱质谱联用仪(日本岛津公司);ACQUITY UPLC I-Class 系统(美国Waters 公司);TripleTOFTM5600 高分辨四极杆-飞行时间质谱仪(美国AB Sciex 公司),配备电喷雾离子源(electro⁃spray ionization,ESI);AVANCE Ⅲ400 MHz 核磁共振波谱仪(德国Bruker 公司);1200 制备液相色谱仪(美国Agilent 公司);RV 10 旋转蒸发仪(德国IKA 公司);KQ-600DE 数控超声波清洗器(昆山市超声仪器有限公司)。
甲醇、乙腈、甲酸(色谱纯,德国Merck 公司),氘化三氯甲烷(CDCl3,99.8%;美国剑桥同位素实验室),Milli-Q Advantage A10 超纯水系统(美国Millipore公司)。
1.2 仪器条件
1.2.1 制备液相分析条件
使用ZORBAX SB-C18色谱柱(250 mm×21.2 mm,7 μm;美国Agilent 公司),柱温为室温,流动相采用V水∶V甲醇=15∶85的甲醇水溶液,分析时间为7 min,流速为20 mL/min,进样体积为200 μL,采样波长为210 nm。根据出峰时间,收集4.8~5.5 min时间段的洗脱液。
1.2.2 GC-MS分析条件
使用DB-5MS 石英毛细管柱(30 m×0.25 mm,0.25 μm;美国Agilent 公司)。升温程序:140 ℃保持3 min,以20 ℃/min 升至320 ℃,保持13 min。载气为氦气,流速为1 mL/min,分流比为40∶1,溶剂延迟3 min,进样口温度280 ℃,电子轰击(electron impact,EI)离子源,电子能量为70 eV,离子源温度为230 ℃,传输线温度为250 ℃,质量扫描范围为m/z35~500。
1.2.3 UPLC-QTOF-MS分析条件
使用Kinetex Biphenyl 色谱柱(100 mm×3.0 mm,2.6 μm;美国Phenomenex 公司),柱温40 ℃,流动相A为V甲酸∶V水=1∶1 000 的甲酸水溶液,流动相B 为乙腈。梯度洗脱程序:初始为0% B;0~12.0 min,0%~100%B;12.0~13.0 min,100% B;13.0~13.1 min,100%~0%B;13.1~15.0 min,0% B。流速为0.8 mL/min;进样量为1 μL;电喷雾离子化-碰撞诱导解离(electrospray ionization-collision induced dissociation,ESI-CID),正离子全扫描及二级质谱扫描;电离喷雾电压5.5 kV,去簇电压80 V;喷雾温度为600 ℃;雾化气(N2)50 psi,脱溶剂气(N2)30 psi,气帘气(N2)30 psi;一级质谱碰撞能量为10 V,质量扫描范围为m/z100~1 000;二级质谱碰撞能量为(35±15)V,质量扫描范围为m/z50~1 000。
1.2.4 NMR分析条件
利用标准实验模板,分别采集样品的一维和二维NMR谱图,分别为1H-NMR、13C-NMR、13C-无畸变极化转移增强(13C-distortionless enhancement by polariza⁃tion transfer,13C-DEPT)、二维核磁共振谱(two-dimen⁃sional nuclear magnetic resonance spectrum,2D NMR)、氢/氢相关谱(1H/1H-correlation spectroscopy,1H/1HCOSY)、碳/氢异核单量子相关谱(1H/13C-heteronuclear single quantum correlation spectroscopy,1H/13C-HSQC)和异核多键碳氢相关谱(1H/13C heteronuclear multi⁃ple bond correlation spectroscopy,1H/13C-HMBC)。1HNMR、13C-NMR 谱的化学位移参考四甲基硅烷(tetra⁃methylsilane,TMS)。
1.3 样品处理
称取50 mg 棕黄色植物状样品(图1),加入1 mL甲醇,超声5 min,以5 000×g离心5 min,取上清液供GC-MS分析。

图1 草本混合物样品照片Fig.1 Photo of the herbal blend sample
称取棕黄色植物状样品约2 g,加入10 mL 甲醇,超声10 min,以5 000×g离心5 min,取上清液。再重复3 次,将4 次的上清液合并,旋蒸至剩余约2 mL,得到1 g/mL 的植物提取浓缩液,过0.45 μm 滤膜,供制备液相分析。采用制备液相对1 g/mL 的植物提取浓缩液进行分离,收集目标组分的洗脱液。累计进样,合并洗脱液,将其转移至玻璃茄形瓶中,80 ℃水浴旋蒸至干,再进一步真空干燥箱中干燥48 h,得到制备样品。
称取制备样品约1 mg,溶于1 mL 甲醇,供GC-MS分析。将配制的1 mg/mL 制备样品甲醇溶液,用0.1%甲酸水溶液稀释,配制成1 μg/mL 溶液,供UPLCQTOF-MS分析。
称取制备样品约5 mg,溶解于1 mL 氘化三氯甲烷中,供NMR分析。
2 结果与讨论
2.1 GC-MS检测
采用GC-MS 分析植物样品提取液,样品总离子流色谱图(图2)中保留时间为12.77 min 的峰为主峰,其EI 质谱图(图3)中存在氟戊基吲哚酰胺类合成大麻素的典型特征离子m/z232 和144,但与NIST 谱库及实验室自建NPS 谱库检索比对无匹配结果。进一步结合《新精神活性物质分析手册:质谱分册》[4]中吲哚酰胺类合成大麻素的质谱特征推测,该物质可能为2-[1-(5-氟戊基)-1H-吲哚-3-甲酰氨基]-3,3-二甲基丁酸甲酯(5F-MDMB-PICA)的乙酯衍生物2-[1-(5-氟戊基)-1H-吲哚-3-甲酰氨基]-3,3-二甲基丁酸乙酯(5F-EDMB-PICA,图4),其可能的EI 裂解途径见图5。EI 质谱图中的基峰m/z232 可能是由化合物酰胺基的C-N 键断裂形成,碎片离子m/z232 的C-N 键进一步断裂形成碎片离子m/z144,碎片离子m/z144 进一步丢失CO 生成了吲哚环m/z116;碎片离子m/z334 可能是由于分子离子丢失C4H8+形成,碎片离子m/z334丢失乙氧基形成碎片离子m/z288,碎片离子m/z288进一步丢失CO生成碎片离子m/z260。

图2 样品的总离子流色谱图Fig.2 Total ion chromatograms of the sample

图3 样品总离子流色谱图中12.77 min组分的EⅠ质谱图Fig.3 EⅠmass spectra of the 12.77 min component in total ion chromatogram of the sample
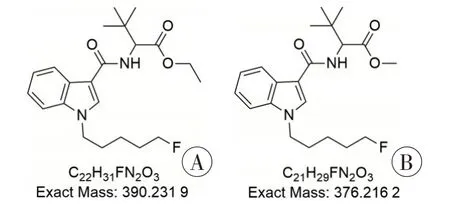
图4 5F-EDMB-PⅠCA、5F-MDMB-PⅠCA的化学结构Fig.4 Chemical structures of 5F-EDMB-PⅠCA and 5F-MDMB-PⅠCA

图5 5F-EDMB-PⅠCA在EⅠ模式下可能的裂解途径Fig.5 Proposed fragmentation pathways of 5F-EDMB-PⅠCA in EⅠmode
2.2 未知化合物的制备
NMR 波谱法是确证物质结构的金标准,本实验室曾鉴定大量未知固体缴获物,最终结构的确证均是通过化合物一维和二维NMR 谱图的解析完成[11-12]。但是对于含量较低的植物制品,NMR 谱图干扰严重,难以直接进行结构解析,因此,先使用制备液相法对目标物进行纯化。在进行未知合成大麻素纯化制备前,先对制备液相法的流动相组成和进样体积这两个条件进行优化。
收集洗脱液,进GC-MS 分析,从而确定其中所包含的成分。实验结果表明,当采用V水∶V甲醇=10∶90、V水∶V甲醇=15∶85、V水∶V甲醇=20∶80 3 种流动相进行分离时,目标合成大麻素的出峰时间分别为3.65、5.08和7.43 min(图6)。综合考虑分离度和分析时间,最终选取的流动相为V水∶V甲醇=15∶85。

图6 不同流动相条件下获得的色谱图Fig.6 Chromatograms acquired using different mobile phases
质量浓度为1 g/mL 的植物提取液在进样体积为50、100、200 μL 时的液相色谱图见图7。结果表明,选用3 种进样体积均可以实现有效分离,为节省分析时间,最终选取的进样体积为200 μL。

图7 不同进样体积条件下获得的色谱图Fig.7 Chromatogram acquired using different injection volumes
在最优化的条件下,采用制备液相对1 g/mL的植物提取浓缩液进行分离,收集5.08 min 的洗脱液。累计进样10次,合并得到约200 mL洗脱液,将其转移至玻璃茄形瓶中,80 ℃水浴旋蒸至干,再进一步真空干燥箱中干燥48 h,得到浅黄色油状物约10 mg。
2.3 未知化合物的结构解析
采用GC-MS 分析制备样品,样品的总离子流色谱图见图2B。制备样品的EI 质谱图(图3B)与植物样品提取液的EI质谱图(图3A)高度一致。
采用UPLC-QTOF-MS 分析制备样品,化合物的出峰时间为10.07 min。在ESI 正离子模式下,化合物的一级质谱图(图8A)中显示其准分子离子[M+H]+为m/z391.239 1,预测分子式为C22H31FN2O3,与5FEDMB-PICA 的分子式吻合。ESI-CID 模式下的二级质谱图(图8B)中有两个碎片离子,m/z232.113 2(C14H15FNO+)和m/z144.044 3(C9H6NO+),这两个碎片离子是氟戊基吲哚酰胺类合成大麻素的典型特征离子[4],其中232.113 2 是由酰胺基的C-N 键断裂形成,该碎片离子的C-N 键进一步断裂形成碎片离子144.044 3。

图8 UPLC-QTOF-MS分析5F-EDMB-PⅠCA的质谱图及可能的质谱裂解途径Fig.8 Mass spectra obtained by UPLC-QTOF-MS for 5F-EDMB-PⅠCA and proposed fragmentation pathways
采用NMR 分析制备样品,其1H-NMR、13C-NMR谱图见图9。根据制备样品的EI 质谱图和ESI 质谱图,大致推断未知物是5F-MDMB-PICA 的乙酯衍生物5F-EDMB-PICA。5F-EDMB-PICA 与5F-MDMBPICA 的结构差异仅为1 个CH2,将两者碳谱中谱峰的化学位移进行比较(表1),结果显示,两者C-2 至C-23 之间所有碳的化学位移几乎一致,证实了两个化合物具有相同的2-[1-(5-氟戊基)-1H-吲哚-3-甲酰氨基]-3,3-二甲基丁酸结构。5F-EDMB-PICA 的H-26(δ=4.23)氢的个数为2,H-27(δ=1.32)氢的个数为3;H-26(δ=4.23)与C-23(δ=172.3)、C-27(δ=14.4)存在1H/13C-HMBC 耦合,H-27(δ=1.32)与C-26(δ=61.2)存在1H/13C-HMBC耦合,证实了5F-EDMB-PICA中的丁酸乙酯结构。

图9 5F-EDMB-PICA 的1H-NMR和13C-NMR谱图Fig.9 1H-NMR and13C-NMR spectra of 5F-EDMB-PⅠCA
综合GC-MS、UPLC-QTOF-MS 和NMR 的检验结果,确定未知物合成大麻素为5F-EDMB-PICA。5FEDMB-PICA 的1H-NMR、13C-NMR 谱中各谱峰的详细归属见表1。5F-EDMB-PICA 是已知化合物5FMDMB-PICA 的乙酯衍生物,两个化合物的结构仅差一个甲基,同属于氟戊基吲哚酰胺-丁酸酯结构的合成大麻素。5F-MDMB-PICA 是2019—2020年本实验室检出频率最高的合成大麻素类物质,于2021年7月1 日起被列入《非药用类麻醉药品和精神药品管制品种增补目录》。

表1 5F-EDMB-PⅠCA的1H-NMR、13C-NMR数据Tab.1 1H-NMR and13C-NMR data of 5F-EDMB-PICA

续表1Continued Tab.1
3 结论
本研究以一份送检合成大麻素植物制品检材为例,详细介绍了无标准品时植物制品中未知合成大麻素的定性分析策略。采用制备液相法从植物制品提取液中分离、纯化合成大麻素,并综合利用GC-MS、UPLC-QTOF-MS、NMR 实现对未知合成大麻素结构的解析,同时还对5F-EDMB-PICA 在EI 和ESI-CID两种模式下生成的碎片离子进行了裂解途径的推导,对5F-EDMB-PICA 的1H-NMR、13C-NMR 谱的谱峰进行了解析和归属,这些信息将有助于法庭科学实验室在案件中鉴定该物质或其他具有类似结构的化合物。对于已知的合成大麻素,传统的质谱定性方法通常是使用标准物质,将目标化合物的色谱保留时间和质谱图与标准物质进行比较,以确定化合物的结构。然而,在NPS 新化合物数量快速增长的情况下,很难及时获取标准物质,且新化合物的质谱图也很难被添加到通用的商业化质谱库中。如果没有标准物质,则需要综合利用质谱、核磁等进行化学结构的解析,而NMR 波谱的分析需要一定量的纯化合物。采用化学合成法合成化合物成本高、周期长,制备液相法简单、快速、高效,为解决NPS 制品中低含量未知化合物的定性分析难题提供了备选方案。
猜你喜欢
当代水产(2022年4期)2022-06-05
口腔护理用品工业(2021年4期)2021-11-02
环境保护与循环经济(2021年7期)2021-11-02
山东第一医科大学(山东省医学科学院)学报(2021年7期)2021-10-13
食品安全导刊(2021年20期)2021-08-30
昆明医科大学学报(2021年2期)2021-03-29
昆明医科大学学报(2020年12期)2021-01-26
山东化工(2019年11期)2019-06-26
儿童故事画报·发现号趣味百科(2017年1期)2017-06-01
华声(2016年20期)2016-11-19
