
荷花腐败病菌的荧光定量PCR检测
2021-12-13 08:52温华强舒灿伟曾莉莎周而勋
中国农学通报 2021年34期
温华强,舒灿伟,曾莉莎,周而勋
(1华南农业大学植物保护学院/广东省微生物信号与作物病害防控重点实验室,广州 510642;2东莞市农业科学研究中心,广东 东莞 523086)
0 引言
荷花是一种重要的经济作物,其地下茎(藕)及种子(莲子)均可食用,其花具重要的观赏价值。中国荷花栽培历史悠久,除西藏和青海外,各地均有种植,是世界荷花的栽培中心[1]。近年来,在广东珠三角的荷花栽培集中区爆发了大范围的荷花腐败病,严重制约了荷花产业及旅游业的发展[2]。荷花腐败病又称腐烂病、枯萎病,是荷花种植区发生最严重、最为普遍的病害之一[3]。该病害具有爆发速度快、破坏性强等特征,在整个荷花生长周期内均可发生,如果无有效的防治措施和防治药物,能够造成20%的减产,严重的可达60%以上,甚至绝收[4-5]。2014年,Zhu等[6]鉴定了荸荠枯萎病,认为国内的荸荠枯萎病病原菌为普通镰刀菌(Fusarium commune)及一个未定名的镰孢菌(Fusariumsp.)。2016年,Zhu等[7]建立了检测土壤及植物组织中荸荠枯萎病菌(F.commune)含量的荧光定量PCR检测方法。2017年,曾莉莎等[8]对广东、广西、江西、福建、湖南、湖北等地的荷花腐败病病原菌进行鉴定及系统发育研究,通过形态学和分子鉴定,认为普通镰刀菌(F.commune)是引起荷花腐败病的病原菌,并且与分离自国内荸荠枯萎病的病原菌为同种。
引起荷花腐败病的镰刀菌是一种土壤习居菌,其菌丝体和厚垣孢子可以在土壤长期存活,成为来年的再侵染源[9]。荷花为多年生水生植物,荷花腐败病的发病程度与水底土壤中的荷花腐败病菌含量密切相关。对此,掌握荷花腐败病发生程度和土壤中荷花腐败病菌的数量的关系,将有助于在荷花种植前后有针对性地防治该病害。然而,传统的检测镰刀菌的方法,比如平板稀释法,对其进行分离、形态学以及致病性鉴定等,步骤繁琐、耗时较长,当病害爆发时,由于诊断缓慢,往往延误最佳防治时机,很容易造成严重的损失[10-12];常规PCR相对来说速度较快、特异性高,但检测灵敏度仍有待提高[13];对土壤中的病原菌进行实时定量检测可为制定科学的病情防控方案提供更加科学的依据[14]。本研究以此为切入点,联合东莞市香蕉蔬菜研究所技术人员对该病害的发展动态进行监测,旨在研发一种快速、准确的定量检测荷花腐败病菌的荧光定量PCR技术,对东莞莲湖的土壤、莲藕等样品的荷花腐败病菌数量进行动态监测,旨在为该病害的有效防治提供重要依据。
1 材料与方法
1.1 荷花腐败病样品的采集与病原菌分离
从广东省东莞市桥头镇莲湖的荷花腐败病发病区采集病藕组织,通过常规分离、培养、纯化和鉴定,获得荷花腐败病菌(F.commune)纯净菌株,用于本研究的所有实验。
1.2 荷花腐败病区土壤中的荷花腐败病菌基因组DNA的提取
土壤的基因组DNA使用E.Z.N.A.®Soil DNA kit(Omega Bio-Tek,USA)试剂盒提取。
1.3 荷花腐败病菌菌丝基因组DNA的提取
将荷花腐败病菌菌株接种于200 mL的PDB培养基,置于25℃、180 r/min条件下的摇床上培养5~8天。将摇床培养的PDB培养基取出,用抽滤机抽滤,得到荷花腐败病菌的菌块(含菌丝和孢子),用灭菌的滤纸吸干水分,置于-80℃下保存。荷花腐败病菌DNA的提取使用E.Z.N.A.®High Performance(HP)Fungal DNA Kit(Omega Bio-Tek,USA)试剂盒。
1.4 各种发病级别荷花病组织基因组DNA的提取
各种发病级别荷花组织基因组DNA的提取使用E.Z.N.A.®High Performance(HP)Plant DNA(Omega Bio-Tek,USA)试剂盒。
1.5 qPCR引物的选用
由于本研究的荷花腐败病菌与国内报道的荸荠枯萎病菌均为普通镰刀菌(F.commune)[8,12],本研究采用的引物是朱志贤[15]筛选的、对荸荠枯萎病菌具有高度特异性的一对引物FO1(5’-GCCATAGGTCAGATAAC CAGTT-3’)和 FO2(5’-TCACTACTGGTGTCAGAAA CGG-3’),这对引物由北京擎科新业生物技术有限公司合成。
1.6 qPCR标准曲线的制作及灵敏度检测
1.6.1 qPCR扩增的体系及使用材料和仪器 实验的qPCR采用SYBR Green I荧光染料,PCR扩增体系为20µL,每份体系包含10.0µL ChamQ Universal SYBR qPCR Master Mix,10 µmol/L(引物工作浓度)的引物各0.4µL(FO1和FO2),1~2µL的DNA模板,加入RNase-free ddH2O至20µL。qPCR扩增的条件如下:95℃预变性 30 s;95℃变性 10 s,60℃延伸 30 s;95℃15 s,60℃ 60 s,95℃ 15 s。进行qPCR反应所用的仪器是Bio-Rad CFX96TM(Bio-Rad Laboratories,CA),获得的数据使用Bio-Rad CFX Manager 3.1(admin)软件进行分析
1.6.2 荷花腐败病菌基因组DNA标准曲线的制作及灵敏度检验 将荷花腐败病菌的基因组DNA稀释至10 ng/µL,再以10倍浓度梯度稀释荷花腐败病菌的基因组DNA,即稀释后的浓度依次为10 ng/µL、1 ng/µL、100 pg/µL、10 pg/µL、1 pg/µL、100 fg/µL、10 fg/µL、1 fg/µL。运行qPCR程序后收集数据,根据Ct值的含义,即每个反应管内的荧光信号到达设定的域值时所经历的循环数,认为具有Ct值的为阳性样本,没有Ct值的是为阴性样本,灵敏度为与阴性样本相邻的最后一个阳性样本的浓度值。
1.6.3 加入植物组织DNA后荷花腐败病菌基因组DNA标准曲线的绘制及灵敏度检测 为研究在自然状态下,荷花腐败病菌侵染健康的荷花组织时,荷花的基因组DNA是否会对荷花腐败病菌DNA的扩增造成影响,实验中提取健康的荷花基因组DNA,稀释至10 ng/µL,再吸取1µL加入到上述10倍浓度梯度的荷花腐败病菌的基因组DNA配的20µL qPCR体系中(1µL荷花腐败病菌的基因组DNA模板和1µL健康的荷花组织基因组DNA)进行扩增,绘制其标准曲线运行qPCR程序后收集数据,根据Ct值的含义,即每个反应管内的荧光信号到达设定的域值时所经历的循环数,认为具有Ct值的为阳性样本,没有Ct值的是为阴性样本,灵敏度为与阴性样本相邻的最后一个阳性样本的浓度值。
1.6.4 接种土壤中荷花腐败病菌标准曲线的绘制及灵敏度检测 将荷花腐败病菌菌株接种于200 mL的PDB培养基,置于25℃、180 r/min条件下的摇床上培养5~8天。准备灭过菌的纱布,用6层灭过菌的纱布过滤出荷花腐败病菌的孢子,并将过滤好的孢子悬液转移至15 mL离心管,10000×g离心15 min,用去离子灭菌水重复洗涤2~4次,至悬液颜色由黄色变为无色(或接近无色)。将分生孢子悬液置于显微镜下,利用血球板计数法,加入去离子蒸馏水稀释成4×107、4×106、4×105、4×104、4×103、4×102、4×101、4个/mL等10倍浓度梯度的孢子悬液。将采自东莞桥头镇莲湖的发病区土壤样本去除杂物,置于干热灭菌箱中,90℃、5 h灭菌烘干3次至恒重,利用研钵将其磨碎,称取800 mg的灭菌土壤至15 mL离心管中,依次将一系列浓度梯度的分生孢子悬液取800µL加至上述的15 mL离心管中,以未接种孢子悬液的灭菌土壤作空白对照,使用E.Z.N.A.®Soil DNA kit(Omega Bio-Tek,USA)试剂盒对其提取基因组DNA,对其进行qPCR扩增和绘制标准曲线,认为具有Ct值的为阳性样本,没有Ct值的是为阴性样本,灵敏度为与阴性样本相邻的最后一个阳性样本的浓度值。
1.7 不同发病级别荷花植株中荷花腐败病菌的qPCR检测
在东莞桥头莲湖采集依次为无病、轻度发病、重度发病3个发病级别的荷花植株(病情分级标准由东莞桥头工作人员制定)各3株,利用随机取样的方法在各种不同的发病级别的荷花植株中取3支茎秆,用5%的次氯酸钠将其浸泡消毒5 min,接着用去离子灭菌水冲洗2~4次,用灭菌的吸水纸吸干表面水分。用灭菌处理过的剪刀依次从3支茎秆中部剪取一小段约100 mg的茎秆组织,提取其基因组DNA,进行qPCR扩增来检测各种发病级别的荷花植株中荷花腐败病菌的含量(根据测得的DNA含量计算),3次平行重复实验。各种发病级别荷花植株的茎秆中的荷花腐败病菌基因组DNA含量,是根据加入10 ng健康荷花组织DNA绘制的10倍浓度梯度的荷花腐败病菌的基因组DNA的标准曲线获得。
1.8 不同发病级别荷花植株根部土壤中荷花腐败病菌的qPCR检测
从各种发病级别的荷花植株的根部土壤处采集3份土壤样品,每份大约1 kg。先将土壤样品去除杂质,再将其置于干热灭菌箱中,打开鼓风功能,38℃、24 h风干。提取处理后的土壤样品的DNA,再进行qPCR扩增,得到相应的Ct值,根据接种荷花腐败病菌的土壤中的荷花腐败病菌基因组DNA的标准曲线,得到各种发病级别荷花植株的根部土壤中荷花腐败病菌的孢子浓度。
1.9 数据分析
每一次的qPCR扩增反应由Bio-Rad CFX Manager 3.1(admin)软件计算分析给出Ct值。10倍浓度梯度的荷花腐败病菌的基因组DNA的标准曲线、加入植物组织DNA后荷花腐败病菌基因组DNA的标准曲线和接种荷花腐败病菌的土壤中的荷花腐败病菌基因组DNA的标准曲线用Microsoft Excel(office 2016)软件制作。用SPSS 22.0软件的协方差分析检验,对是否添加健康荷花组织基因组DNA的一系列浓度梯度的荷花腐败病菌的基因组DNA制作的2条标准曲线之间的截距,以及斜率是否存在显著性差异。用相关分析方法分析各种发病级别的荷花植株中包含的荷花腐败病菌的基因组DNA,以及各种发病级别荷花植株的根部土壤中包含的荷花腐败病菌的基因组DNA与它们的发病级别之间的关联。
2 结果与分析
2.1 qPCR的标准曲线及检测灵敏度
用一系列浓度梯度的荷花腐败病菌基因组DNA进行Real-Time PCR反应,得到的反应曲线如图1所示。利用Microsoft Excel软件制作的荷花腐败病菌基因组DNA的标准曲线(图2)为Y=-4.2734X+36.892(R2=0.9977),其中横坐标X表示稀释的一系列浓度梯度的DNA的浓度的对数,纵坐标Y表示与每个DNA浓度X值相对应的Ct值,R2=0.9977表明该标准曲线具有良好的线性关系。通过对qPCR扩增曲线(图1)分析可得,qPCR的最低检测限度为1 fg/µL。对该qPCR反应的溶解曲线(图3)进行分析,扩增的产物仅有一个峰值(78.5℃),表明该qPCR扩增的DNA产物具有高度的特异性。
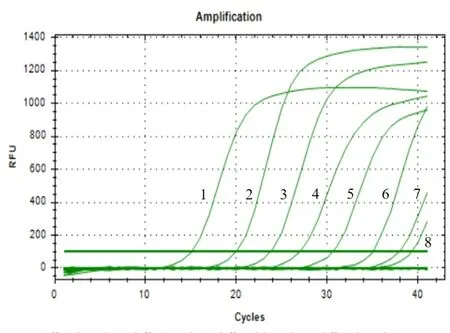
图1 10倍浓度梯度的荷花腐败病菌基因组DNA的qPCR扩增曲线
2.2 加入植物组织DNA后荷花腐败病菌基因组DNA的标准曲线及检测灵敏度
在添加健康荷花组织的基因组DNA后制作的一系列浓度梯度荷花腐败病菌的DNA标准曲线(图2)为Y=-3.6419X+34.501(R2=0.9939)。对添加了健康荷花组织的基因组DNA后的qPCR进行最低检测限度分析,该qPCR反应的最低检测限度为10 pg/μL。利用协方差分析检验2条标准曲线发现截距的差异性不显著(P>0.05),但当比较斜率时则发现2条曲线的差异性显著(P=0.01<0.05)。造成以上2条标准曲线之间存在差异的因素在于添加健康的荷花组织的基因组DNA,故采用添加了健康荷花组织的基因组DNA后制成的标准曲线计算发病的荷花组织中的荷花腐败病菌的DNA含量。
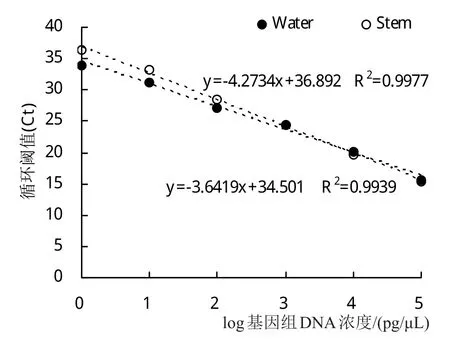
图2 SYBR Green I PCR扩增基因组标准曲线
2.3 接种土壤中荷花腐败病菌的标准曲线及灵敏度检测
提取浓度依次为4×107、4×106、4×105、4×104、4×103、4×102、4×101、4、0 个分生孢子/(g·土)的基因组DNA进行qPCR反应,没有接种孢子的灭菌土壤为对照组(图4)。制成的标准曲线中坐标轴X表示分生孢子在土壤中的数目的对数,坐标轴Y表示其对应的Ct值,两者的线性关系为Y=-3.1995X+34.262(R2=0.9911)(图5)。由图4可知,当分生孢子浓度低于4×102个分生孢子/(g·土)时不能被检测出来,即最低检测限度为4×102个分生孢子/(g·土)。
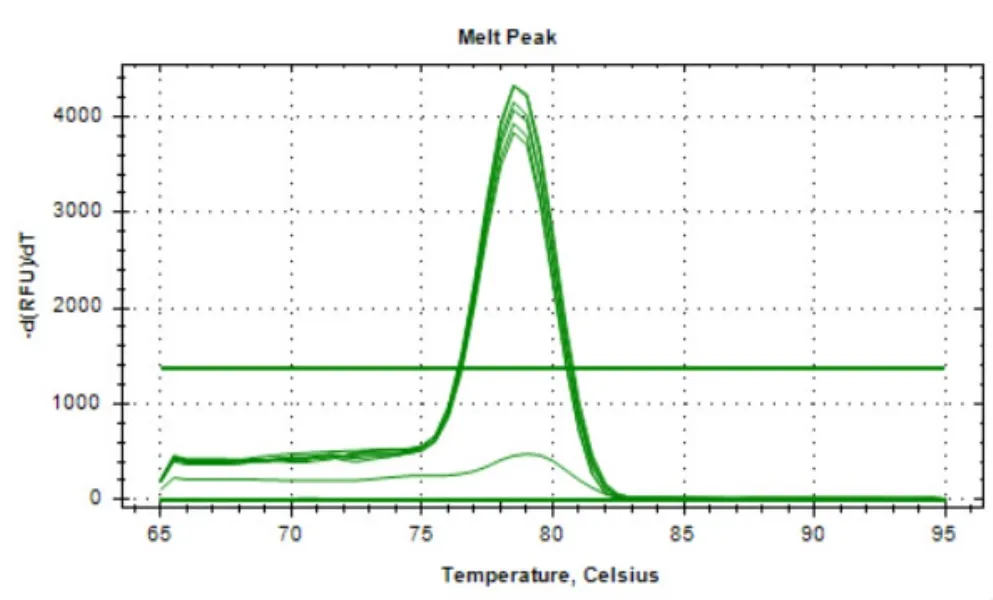
图3 SYBR Green I PCR熔解曲线
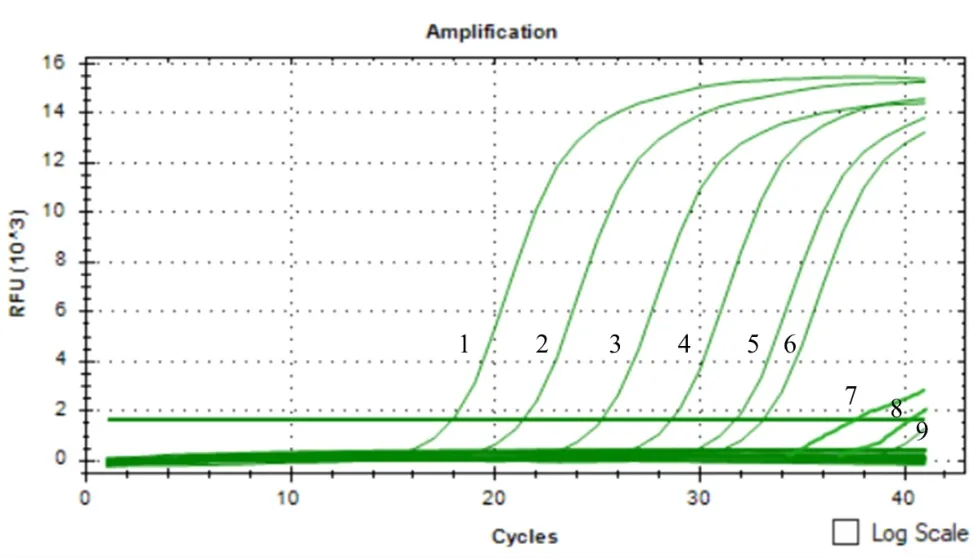
图4 接种土壤的SYBR Green I PCR扩增曲线
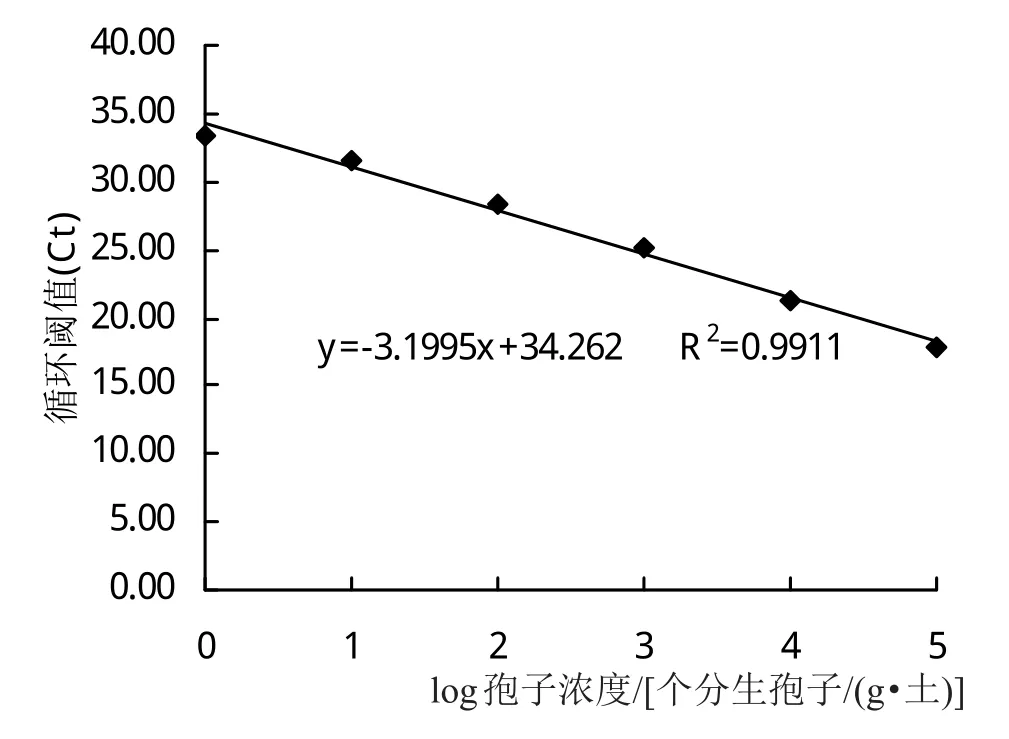
图5 接种荷花腐败病菌的土壤中的荷花腐败病菌基因组DNA的标准曲线
2.4 各种发病级别荷花植株中荷花腐败病菌含量与病害发生程度的相关性
用qPCR方法对各种发病级别的荷花植株中荷花腐败病菌的含量进行了检测。数据显示,各种发病级别荷花植株样品中荷花腐败病菌DNA的含量依次为(0.010±0.003)、(0.0827±0.053)、(1.432±0.439)ng(表1)。其中,2级的DNA含量为0级的143.2倍。染病植株中荷花腐败病菌DNA的含量与其发病程度呈正相关,皮尔森相关系数为0.927,显著性(双尾)为0.044。
2.5 各种发病级别植株地下土壤中荷花腐败病菌含量与病害发生程度的相关性
0~2级发病级别植株的地下土壤中荷花腐败病菌的含量依次为(3.618±2.284)×103、(34.400±12.891)×103、(164.733±54.270)×103个分生孢子/(g·土)。皮尔斯相关系数为0.819,显著性(双尾)为0.389,表明发病的荷花植株地下土壤荷花腐败病菌的含量与病害发生程度呈正相关(相关度高),但显著性较低。
3 结论与讨论
随着人们对荷花的观赏需求和食用需求日益增加,荷花产业发展前景好。然而,素有“荷花癌症”之称的荷花腐败病却成了荷花产业发展的最大障碍之一,这种土传性镰刀菌病害侵染源是病残体、病土等携带的病菌,包括菌丝体、厚垣孢子等镰刀菌的越冬形态。据报道,轮作8年的土壤仍有菌丝和孢子能存活下来,并再度侵染健康的植株,所以仍然不能消除该病害的发生[16]。因此,研发一种能够快速、有效地检测土壤和罹病组织中荷花腐败病菌含量的方法,对于荷花产业的健康发展具有重要的指导意义。
本研究利用SYBR Green I qPCR技术检测了感染荷花腐败病的荷花植株及其地下土壤中含有的荷花腐败病菌的数量,其中qPCR方法的最低检测限度是1 fg/μL,添加了健康的荷花植株的DNA后最低检测限度是10 pg/μL,将分生孢子接种到经灭菌处理的土壤后的最低检测限度是400个孢子/(g·土)。存在于植物或土壤中的PCR抑制因子,对qPCR检测具有限制作用[17-18]。相比普通PCR,实时荧光定量PCR具有灵敏度高、精确定量、实时检测扩增产物量的变化等诸多优点,更加适合用来动态监测荷花腐败病菌的含量与发病程度的关系,分析荷花腐败病菌的含量与发病级别的相关性。荷花腐败病的初侵染源是带菌的种藕和土壤,最初的发病部位是地下茎部,不易观察,常失去最佳防治适期[5]。qPCR具有诸多优点,能够在病害发生的初期进行快速检测,而传统的检测手段难以做到,故已被广泛地应用于病原物的快速检测[19-21]。实时荧光定量PCR技术被广泛应用于土壤中镰刀菌引起的植物病害的快速诊断,陈利达等[22]根据镰刀菌属的TEF-1α基因构建的qPCR检测体系,其灵敏度比常规PCR高104倍;王瑜等[23]根据腐皮镰刀菌(F.solani)的tef 1基因,建立了腐皮镰刀菌的SYBR Green qPCR检测技术,其对重组质粒标准样品的检测灵敏度(1.0×102copies/μL)和稳定性(变异系数0.96%~1.68%)均优于常规PCR。李文学等[24]根据GenBank中葡萄霜霉病菌的cox2基因,建立并优化qPCR反应体系,该体系的检测灵敏度是普通PCR检测灵敏度的100倍,可达0.1 pg/μL。 Megan Ceris Matthews 等[25]利 用 SYBR green PCR技术对香蕉枯萎病菌(F.oxysporumf.sp.cubense,Foc)进行检测,结果表明对Foc谱系VI、TR4和STR4的qPCR检测具有重复性和可重复性,其中,LOQ值为10-4~10-3ng/μL,LOD为10-5~10-4ng/μL。
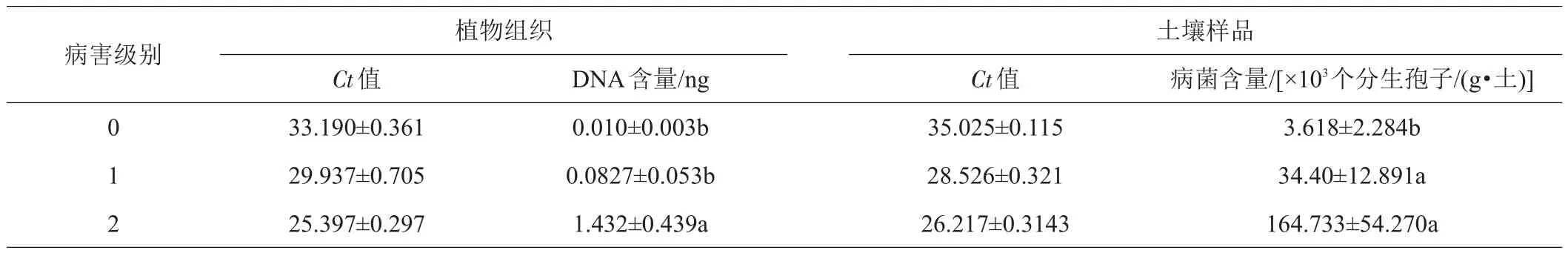
表1 不同病害级别荷花植株中荷花腐败病菌DNA的含量及土壤中荷花腐败病菌的含量
对各种发病级别的荷花地下茎组织及土壤中荷花腐败病菌的含量与发病程度进行相关分析,结果表明,荷花植株中荷花腐败病菌的数量与其发病的程度呈显著的正相关,而土壤中荷花腐败病菌的数量与发病的程度相关性低(无显著的相关性),但荷花腐败病菌的数量还是随着病害发生程度的增加而增加。当然,本研究也存在着一些不足,比如病害的分级偏少、实验样本的处理以及qPCR反应优化问题[26]等。因此,在后续的研究中,还需要建立明确的荷花腐败病病害分级系统,进一步分析土壤或植株中病原菌数量与病害发生程度的关系,从而指导荷花腐败病的防控。综上所述,本研究建立的qPCR定量检测方法是研究此类土传病害强有力的检测手段。
猜你喜欢
基层中医药(2021年8期)2021-11-02
特别健康(2018年3期)2018-07-04
家庭影院技术(2018年5期)2018-06-29
家庭影院技术(2018年3期)2018-05-09
校园英语·下旬(2017年6期)2017-07-14
中学生(2017年13期)2017-06-15
小天使·一年级语数英综合(2017年6期)2017-06-07
阅读与作文(小学高年级版)(2016年11期)2016-12-10
广东农业科学(2016年2期)2016-07-13
衡阳师范学院学报(2016年3期)2016-07-10
