
基于机器学习的二氧化碳电化学还原制备甲酸盐研究
2022-01-10 03:09刘文萱张嘉毅陆奇张皓晨
化工学报 2021年12期
刘文萱,张嘉毅,陆奇,张皓晨
(清华大学化学工程系,化学工程联合国家重点实验室,北京 100084)
引 言
电化学还原二氧化碳(CO2electrochemical reduction,CO2ER)可以在相对温和的条件下将大气中的二氧化碳转化为高附加值的化学产品,如甲酸盐、乙烯、甲烷等,为缓解温室效应、全球变暖等环境问题提供了一个具有前景的解决方案[1-12]。甲酸盐作为CO2ER的重要产品之一,在皮革鞣制、动物饲料、燃料电池等方面应用广泛,经济效益高,具有良好的商业化前景[5,13-17]。与一氧化碳、甲烷等气态产品相比,甲酸盐作为液态产品,还便于储存和运输[15]。但受目前已有催化剂性能的限制,电化学还原二氧化碳制甲酸盐依然面临着选择性差、能耗高、稳定性低等不足,该过程仍未能满足实际应用的需求,开发新型电催化材料依然是实现CO2ER高效生产甲酸盐的关键。
近年来,科学家们通过实验方法已研制出锡[5,12,15,18]、铅[19]、铋[19]等金属或相应的化合物材料用于CO2ER制甲酸盐,但往往无法同时实现高法拉第效率、高电流密度以及低过电位。特别在长时间电解后,这些材料会出现法拉第效率明显下降、过电势明显上升或电流密度显著降低的问题。为得到各方面性能优异的催化剂,往往需要快速筛选大量具有潜在催化性能的材料。然而通过实验方法研究新型催化材料往往具有研发周期长、需要多次重复测试等缺点,限制了潜在催化材料的高速筛选。目前,材料高通量计算技术的迅速发展为研究新型催化剂提供了许多便利,能有效缩短研发周期,降低研发成本,加快高性能催化材料的筛选过程,已受到学术界越来越多的关注[20-28]。例如,Yang等[23]利用机器学习构建模型,快速筛选了具有潜在催化活性的合金材料用于二氧化碳还原,为催化剂的设计和发展提供了理论基础。而Chen等[24]则通过结合多尺度模拟、量子力学以及机器学习的方法,研究了催化剂表面的活性位点,推动了材料表面活性位点可视化的深入发展。由此可见,理论计算研究在预测催化剂性能、筛选表面活性位点和指导材料设计与合成等方面具有积极的作用。而目前针对二氧化碳电催化还原制备甲酸的高通量催化材料筛选鲜有报道。本文结合密度泛函理论(density functional theory,DFT)计算和机器学习(machine learning,ML),系统地探究由14种过渡金属元素两两组合而形成的105种双金属中心催化剂(dualmetal-site catalysts,DMSCs)催化二氧化碳电还原制甲酸盐的性能。
1 实验方法
1.1 模型构建
本文所使用的石墨烯-N6-M1-M2双金属中心催化剂计算模型(M1-M2)由加入两个金属原子的氮原子修饰单层石墨烯表面构成,其晶胞参数如下:a=12.78 Å,b=12.30 Å,c=25.00 Å,α=90°,β=90°,γ=90°(1Å=0.1 nm)。该DMSCs的模型如图1所示。

图1 石墨烯-N6-M1-M2双金属中心催化剂模型[灰色球为碳原子,淡蓝色球为氮原子,深蓝色球为金属原子(M1和M2)]Fig.1 The structure of the graphene-N6-M1-M2model[where grey,light blue,and dark blue spheres represent C,N,and transition-metal atoms(M1and M2),respectively]
本文选取了十四种已有报道或可能用于二氧化碳电化学还原制甲酸盐的过渡金属元素作为DMSC的组成元素M1和M2。包括7种3d金属:铬(Cr)、锰(Mn)、铁(Fe)、钴(Co)、镍(Ni)、铜(Cu)、锌(Zn);4种4d金属:钌(Ru)、铑(Rh)、钯(Pd)、银(Ag);3种5d金属:铱(Ir)、铂(Pt)、金(Au)。
1.2 密度泛函理论计算
实验过程中的所有理论计算均采用基于广义梯度近似(generalized gradient approximation,GGA)的Perdew-Burke-Ernzerhof(PBE)泛函,计算体系交换相关能。同时采用投影缀加平面波方法(projector augmented wave pseudo-potentials)计算原子实,其截断能设置为400eV。所有计算均采用VASP模 拟 软 件 包(ViennaAb-initiosimulation package)。为加速自洽场的收敛,采用高斯函数展宽法,设置kBT分别为0.1 eV(针对层状模型)和0.01 eV(针对小分子)。而后将所有计算出的电子能量外推至kBT=0,以获得0K条件下的电子能。此外,还使用Monkhorst-Pack方法设置了3×3×1的k点对层状模型的倒易空间进行采样。与此同时,将DMSCs层状模型垂直方向上的真空层厚度设置为25Å,以避免重复单元在此方向上发生相互作用。
1.3 Gibbs自由能变计算
催化反应的选择性往往可以根据不同反应方向的热力学性质进行推测[29]。在二氧化碳电催化还原的条件下,第一步反应中活性位点可能结合CO2、质子及电子生成甲酸中间体(*HCOO),或直接与质子及电子结合生成氢原子(*H)。其中,能量更低、更稳定的中间产物将占据活性位点,继而发生后续的反应步骤,最终得到甲酸盐或氢气。目前学界普遍承认的二氧化碳电还原制备甲酸盐和氢析出反应的反应路径[29-30]如式(1)~式(3)所示。其中,*代表空DMSCs表面,*A表示吸附在DMSCs表面的物种A。

式(1)显示了电解二氧化碳制甲酸盐的主要步骤,其关键步骤为第一步:反应位点结合CO2、质子和电子,形成关键反应中间体*HCOO。式(2)表示发生氢析出反应的可能途径,关键步骤也为第一步:反应位点结合质子和电子生成吸附状态下的氢原子,即关键反应中间体*H。式(3)则表示吸附状态下的氢原子形成H2的可能途径。本文通过比较DMSCs表 面 生 成*HCOO和*H的 吸 附Gibbs自 由能变(ΔG*HCOO和ΔG*H),作为判断这两种中间体稳定性的依据,进一步确定反应发生的方向。本工作计算ΔG*HCOO和ΔG*H的表达式如式(4)和式(5)所示。若ΔG*HCOO更负,说明*HCOO吸附在DMSCs表面更稳定,倾向占据活性位点,有利于反应朝制备甲酸盐的方向发生。相反,若ΔG*H越负,则表明*H吸附在DMSCs表面更稳定,倾向于发生氢析出反应(hydrogen evolution reaction,HER)。因此,通过比较ΔG*HCOO和ΔG*H,便能初步判断DMSCs参与CO2ER的反应活性和选择性,筛选出潜在的可以实现高效催化二氧化碳电还原制甲酸盐的新型材料。
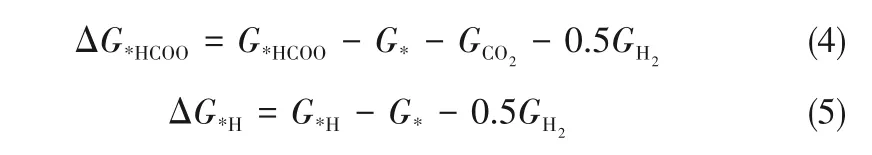
G*HCOO和G*H分 别 为*HCOO和*H的Gibbs自 由能,通过DFT计算得到的体系电子能(E)经修正后求出。修正包含零点振动能(ZPVE)、振动焓和振动熵,计算公式如式(6)所示[29]。G*为空表面催化剂的Gibbs自由能。
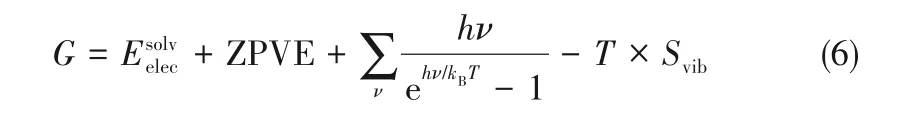
GCO2和GH2也通过DFT计算得到的体系电子能(E)经修正后求出,计算公式如式(7)所示[29]。由于氢气和二氧化碳均为线型分子,此处n取7[29]。
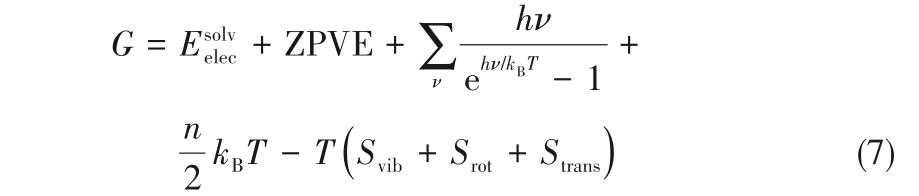
1.4 机器学习
为搭建高效的机器学习模型,本文共选择了Scikit-Learn机器学习库中的8种算法开展研究,分别使用回归模型(regressor)研究和预测ΔG*HCOO、ΔG*H,以及分类模型(classifier)研究和预测*HCOO吸附构型。8种算法具体包括梯度提升(gradient boosting)、K近邻(K-nearest neighbors)、随机森林(random forest)、支 持 向 量 机(support vector machine)、岭回归(ridge)、决策树(decision tree)、AdaBoost算法和Bagging算法。基于8种算法的回归模型缩写分别是GBR、KNR、RFR、SVR、RR、DTR、ABR和BAG;相应的分类模型的缩写为GBC、KNC、RFC、SVC、RC、DTC、ABC和BAGC。
本文根据式(8)计算Pearson相关系数p,对特征之间的相关性开展研究[28]。ai和bi分别代表两种特征的第i个值,aˉ和bˉ代表对应特征的均值。p值分布在1至-1之间,1代表强正相关,-1代表强负相关。p值为0时,表明两种特征无关系。
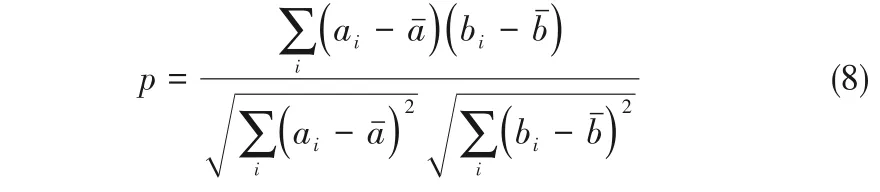
对特征重要度的研究采用基于树结构的GBR模型,通过计算每一种特征在每颗树中重要度的平均值来衡量,计算公式如式(9)所示[28,31-32]。其中,M代表树的数量,(Tm)代表特征j在单棵树中的重要度,具体计算方式见文献[32]。最后采用标准化处理,所有特征重要度的总和为1。

机器学习预测结果的准确性通过均方根误差(RMSE)和决定系数(R2)来判定,计算式如式(10)、式(11)所示[28]。yi、xi分别代表DFT计算结果和机器学习预测结果,yˉ代表DFT计算结果的平均值,n为总次数。R2越接近0,表明该机器学习模型的拟合效果越差;越接近1则表明模型的拟合效果越好。

本文一共选取了十四种过渡金属元素,两两组合后共可得到196种DMSCs。M1和M2在几何层面上是等价的,对于任意一对指定的M1和M2,存在着两个等价的组合,其对应的表面实际为同一个表面。然而由于M1和M2的特征并不完全相同,这两种组合在特征空间内是有显著区别的,因此机器学习研究仍基于196种组合来开展。此外,为降低模型过拟合的风险,同时扩大预测的范围,增加模型的灵活性,本文对DFT所得训练集中的ΔG*HCOO和ΔG*H添加了均值为0、方差为0.03 eV的正态分布噪声[28]。
2 实验结果与讨论
2.1 关键反应中间体*HCOO和*H在DMSCs表面的稳定吸附构型
为得到吸附构型机器学习模型的训练集,本文首先随机选出23种DMSCs进行DFT计算,研究了其表面*HCOO和*H的稳定吸附构型。图2(a)~(c)绘制了23种DMSCs表面*HCOO的三种不同吸附构型。其中,有11种DMSCs采取了如图2(a)所示的M1-*HCOO-M2双配位构型。在该种构型中,*HCOO的两个O原子同时与M1和M2连接。采取双配位构型的M1-M2组合往往具有相似的原子半径,例如Cu-Ni、Ni-Co、Cr-Co和Cr-Mn等。除此之外,若M1和M2具有相似的电子结构,例如Cu-Ag等,也采用了该种构型方式。
偏离M1-*HCOO-M2双配位构型的*HCOO主要存在另外两种吸附构型。图2(b)显示了M-*HCOO单配位构型。在该结构中,一个O原子与M1(或M2)连接,另一个O原子则处于M2(或M1)上方较远位置处,并未与其相键合,共计有8种M1-M2采用该种吸附构型。图2(c)则显示了M-*HCOO单配位扭曲构型。在该结构中,一个O原子与金属M1(或M2)连接,另一个O原子倾向于远离M2(或M1),使整个分子呈现扭曲形态,明显偏离M1-*HCOO-M2双配位构型,采用该种吸附构型的M1-M2共有4种。
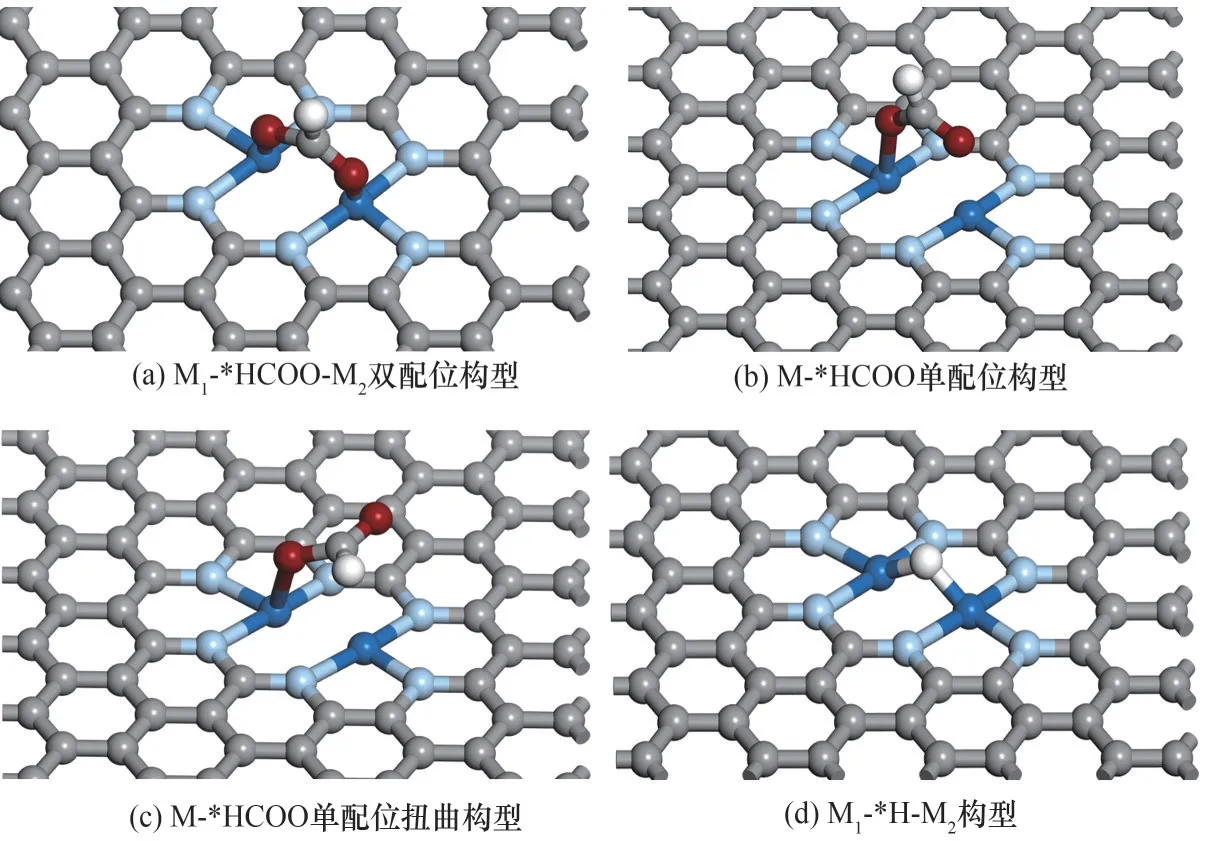
图2 CO2ER和HER关键反应中间体的不同吸附构型[灰色球为碳原子,淡蓝色球为氮原子,白色球为氢原子,红色球为氧原子,深蓝色球为金属原子(M1和M2)]Fig.2 Different adsorption configurations of key intermediates of CO2ER and HER[where grey,light blue,white,red and dark blue spheres represent C,N,H,O and transition-metal atoms(M1and M2),respectively]
对于*H,23种DMSCs均采取相同的吸附方式,*H与两个金属原子同时成键,即*H以二配位形式桥接在M1和M2之间,呈现M1-*H-M2构型,如图2(d)所示。
2.2 Gibbs自由能变计算结果分析
ΔG*HCOO和ΔG*H是决定二氧化碳还原选择性的关键,因此本文计算了上述23种DMSCs的ΔG*HCOO和ΔG*H,作为训练集参与后续的机器学习研究和预测(表1)。计算结果显示,在23种DMSCs中,共有5种组合的ΔG*HCOO小于ΔG*H(*HCOO吸附强,易占据活性位点),表明在这些DMSCs表面*HCOO吸附更稳定,更有利于进行二氧化碳还原。而其余DMSCs的ΔG*HCOO结果均大于ΔG*H(*HCOO吸附弱,不易占据活性位点),表明这些DMSCs的表面更利于形成相对稳定的*H。
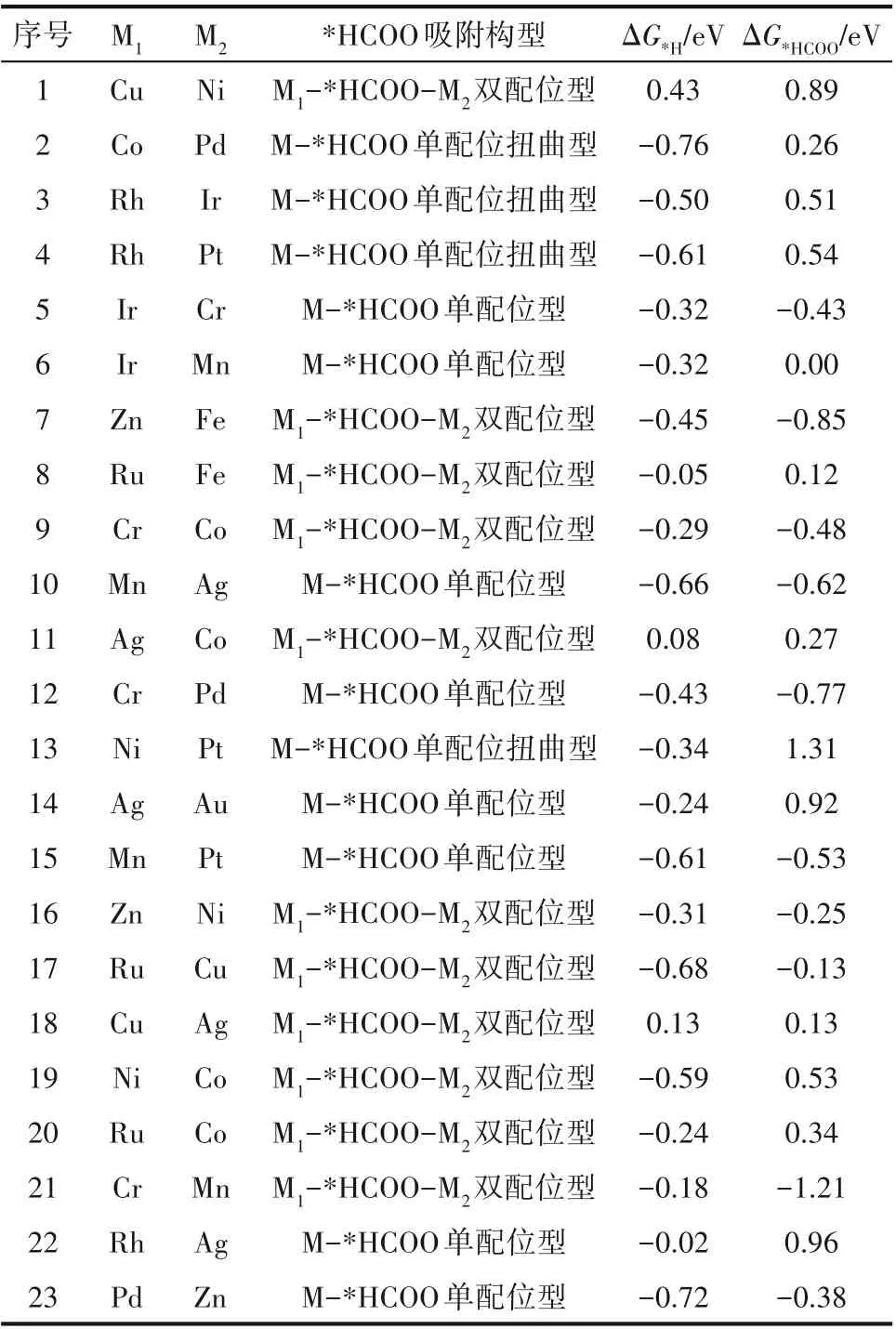
表1 随机抽取的23种DMSCs的ΔG*H、ΔG*HCOO和*HCOO吸附构型Table1 ΔG*H,ΔG*HCOO and*HCOO configurations of23 randomly selected DMSCs
其中,Cr-Mn的ΔG*HCOO值约为-1.21 eV,是所有ΔG*HCOO的最小值。与之相反,Ni-Pt具有最大的ΔG*HCOO,约为1.31 eV。这与吸附构型的计算结果是吻合的。Cr-Mn表面吸附的*HCOO采取了相对稳定的M1-*HCOO-M2双配位构型,对*HCOO的吸附性能更好,促进CO2ER制甲酸盐选择性的提升。而*HCOO在Ni-Pt表面上主要采取单配位扭曲型结构。受该种构型影响,*HCOO与Ni-Pt的相互作用比Cr-Mn更弱,导致ΔG*HCOO更大,使反应不利于生成*HCOO,反而利于形成*H(Ni-Pt表面的ΔG*H为-0.34 eV)。
与Cr-Mn组合类似,其他采取M1-*HCOO-M2双配位构型的部分DMSCs(Zn-Fe和Cr-Co),也展现出较好的*HCOO吸附性能;而剩余采取双配位构型的金属组合的ΔG*HCOO则高于ΔG*H。与Ni-Pt一样采取单配位扭曲构型的Co-Pd、Rh-Ir和Rh-Pt均具有较大的ΔG*HCOO,分别是0.26 、0.51 和0.54 eV。相比较生成甲酸盐,这些组合更有利于生成*H(ΔG*H分别为-0.76 、-0.50 和-0.61 eV)。对于采取M-*HCOO单配 位构 型 的DMSCs来说,仅有Ir-Cr和Cr-Pd两种组合因其具有更小的ΔG*HCOO值,表现出较好的*HCOO吸附性质;而采取该种构型的其余DMSCs的ΔG*H均低于ΔG*HCOO。
此外,还发现23种DMSCs中,所有包含Cr原子的催化剂均有利于生成*HCOO(ΔG*HCOO<ΔG*H)。相比较结合*H,由Cr原子组成的DMSCs更倾向于结合*HCOO,对*HCOO的吸附性能更好。这可能是由于Cr原子具有较好的亲氧性,能够有效地与*HCOO中的氧原子进行配位[33]。
2.3 机器学习模型特征捕捉、筛选和模型优化
完成对23种DMSCs的DFT计算后,本文分别收集了196种DMSCs所含金属原子的14个电子特征和4个几何特征,作为机器学习所需的特征用于描述对应的DMSCs。为进一步提高模型的预测精度,还根据这18个独立特征,增加了34个可通过简单数学运算得出的组合特征[28]。所有的52个特征汇总见表2。此外,可以通过交换M1和M2的位置,将由随机选出的23种DMSCs组成的训练集扩展至46种DMSCs组成的训练集,占全部数据的23.5%。需要说明的是,交换M1和M2的位置得到的新数据点在特征空间内并不与原数据重合,因此可以认为是两个相互独立的数据点。

表2 机器学习的完整52个特征Table2 Complete feature space of52features for ML
过多的特征反而会导致机器学习的训练效率低下,同时影响预测的准确度[20]。为确定最合适的特征数量,本文研究了不同大小的特征空间下GBR对于ΔG*HCOO、ΔG*H以及GBC对于*HCOO吸附构型的机器学习性能。对每一个特征空间下的GBR/GBC模型,采用十重交叉验证法测试其学习性能,即将训练集随机分成十份,每次取一份作为验证数据,剩下九份作为训练数据,使用不同大小特征空间的GBR/GBC模型对训练数据进行学习,比较预测结果与验证数据之间的误差,确定最合适的特征空间大小。为进一步避免随机等分过程引入的误差,还将上述过程重复5次,最终得到50次误差的平均值[28]。
对于ΔG*HCOO,图3(a)显示当不断删去低重要度和相互之间高相关性的特征后,RMSE逐渐降低,学习时间不断减少。当特征数量降低至7时,RMSE取到最小值0.075 eV,学习时间也保持在较低水平。而当进一步减少特征数量时,RMSE反而开始增加,说明特征数量低于7时不足以精确描述体系。因此本文最终筛选出7个特征参与到后续ΔG*HCOO的预测研究中,分别是(Z1+Z2)/2、(N1+N2)/2、(PE1+PE2)/2、(IE1+IE2)/2、(EA1+EA2)/2、(Nd1+Nd2)/2和(WF1+WF2)/2,均与金属原子的电子特征有关。这些电子特征往往会影响二氧化碳还原中间体在DMSCs表面的相互作用强度,从而影响ΔG*HCOO[20]。图3(b)显示了这7个特征在预测ΔG*HCOO时的重要度。其中,重要度最高的是平均原子序数,它的重要度高达0.53 。此外,图3(c)显示的Pearson相关系数也表明所筛选的7个特征之间相关度较低,彼此之间相互独立。
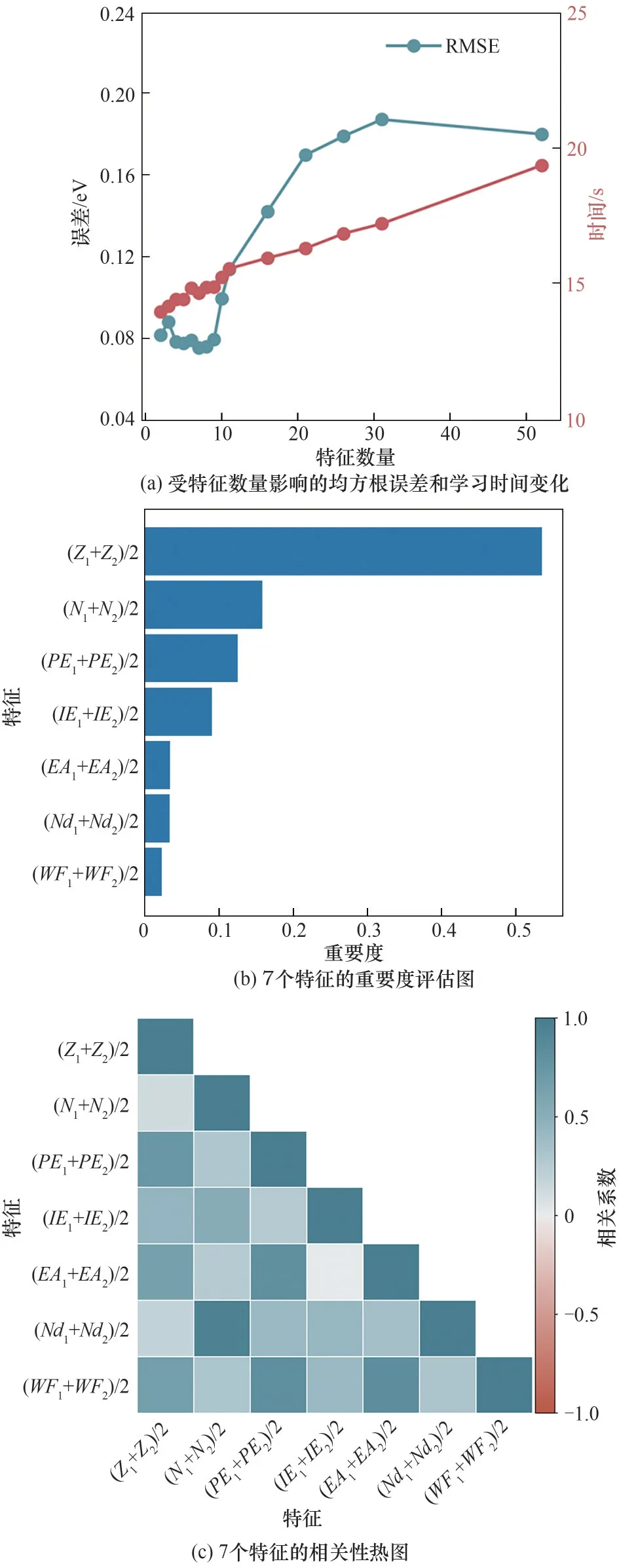
图3 ΔG*HCOO的特征筛选研究Fig.3 Feature selection forΔG*HCOO
与研究ΔG*HCOO的方法类似,本文在研究ΔG*H时进行了相同的特征筛选过程,最终保留了4个特征,分别是(N1+N2)/2、(IE1+IE2)/2、(Nd1+Nd2)/2和(R1+R2)/2,前3个为电子特征,最后1个为几何特征。综合来看,相比于几何特征,DMSCs的电子特征对ΔG*HCOO和ΔG*H的影响更大。对于*HCOO吸附构型,最终共筛选出14个特征,由7个电子特征和7个几何特征组成,分别为PE1-PE2、r1+r2、WF1+WF2、R1+R2、(IE1+IE2)/2、R1、R2、EA1、(R1+R2)/2、Nd1、Nd2、EA2、r1和r2。其中,电负性之差PE1-PE2的重要度最大,占比达0.41。在以往的DMSCs研究中,两个金属原子的电负性差异对DMSCs表面吸附物的吸附强度有显著的影响[20]。当两原子的电负性差距较大时,DMSCs表面的电荷可能会重新分布,更有利于中间体的吸附,这与所筛选出电负性之差为最重要的特征是相吻合的。
除了确定最合适的特征空间大小,本文还对比了8种不同算法的预测精度。在预测ΔG*HCOO时,GBR和DTR模型都展现出较好的拟合效果(图4)。其中,GBR模型预测ΔG*HCOO结果的误差最小(RMSE=0.075 eV),其决定系数也最高(R2=0.999 )。同样地,GBR模型对ΔG*H的预测结果也具有最小的误差(RMSE=0.062 eV)和最高的决定系数(R2=0.995)。在预测*HCOO吸附构型时,GBC显示出了最大的预测准确率,超过80%;而DTC的预测准确率仅有75.5%(图5)。因此本文选择了拟合效果最优的梯度提升模型(GBR和GBC)来开展后续的机器学习预测。
在对剩余150种DMSCs展开预测之前,本文还验证了随机选出的46种DMSCs组成的训练集是否对全数据集(由196种DMSCs组成)具有很好的代表性,以确保基于训练集所训练好的机器学习模型能很好地应用于全数据集。图6展示了研究ΔG*HCOO时筛选出的7个特征在训练集及全数据集下的分布形状。从图6可以看出,每个特征在训练集及全数据集下不同的数值出现的频率相似,说明该特征在训练集和全数据集下的数据值分布情况相近,表明之前随机选出的DMSCs组成的训练集对全数据集具有很好的代表性,基于训练集优化的机器学习模型预测ΔG*HCOO的结果更加可靠[28]。此外,对于ΔG*H和*HCOO的吸附构型,也研究了它们各自对应的特征在训练集和全数据集下的统计分布对比,所得的统计分布情况也相似。
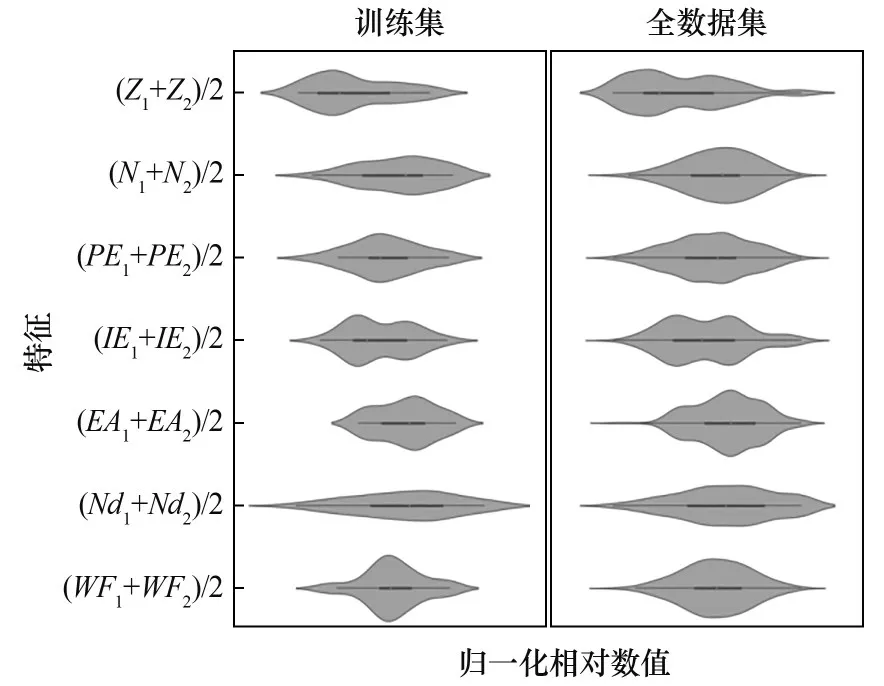
图6 ΔG*HCOO的7个特征在训练集和全数据集下的归一化相对数值的出现频率分布情况Fig.6 Statistical distributions of7features forΔG*HCOO in training data set and full data set
2.4 机器学习预测DMSCs反应性能
基于训练集对机器学习模型的有效训练,本文快速预测了其余150种DMSCs的ΔG*HCOO、ΔG*H和*HCOO吸附构型。对于某一对M1、M2,由于交换M1和M2所获得两种DMSCs的结果差别很小(小于0.01 eV),取二者的平均值,共得到105个唯一的DMSCs组合的预测值。为避免单次预测带来的偶然误差,还将上述预测过程重复5次取平均值,最终结果如图7~图9所示。
图7为105种DMSCs表面ΔG*HCOO的预测结果,预测的ΔG*HCOO分布在-1.21 ~1.31 eV之间。大多数由Mn、Cr、Zn、Fe等 第 四 周 期 金 属 原 子 组 成 的DMSCs的ΔG*HCOO均小于0,十分利于*HCOO中间体的吸附,如图7(a)中的蓝色所示。图7(b)展示了每种金属原子对应的所有DMSCs的ΔG*HCOO平均值。可以看出,由Mn、Cr、Zn、Fe等金属原子对应的平均ΔG*HCOO都小于0,表明由这些原子组成的DMSCs对*HCOO的吸附比较稳定。与此相反,由Au、Pt、Ir、Pd等第五、第六周期金属原子组成的DMSCs的ΔG*HCOO往往大于0,如图7(a)中的红色所示。这部分金属原子对应的平均ΔG*HCOO都超过0.3 eV。
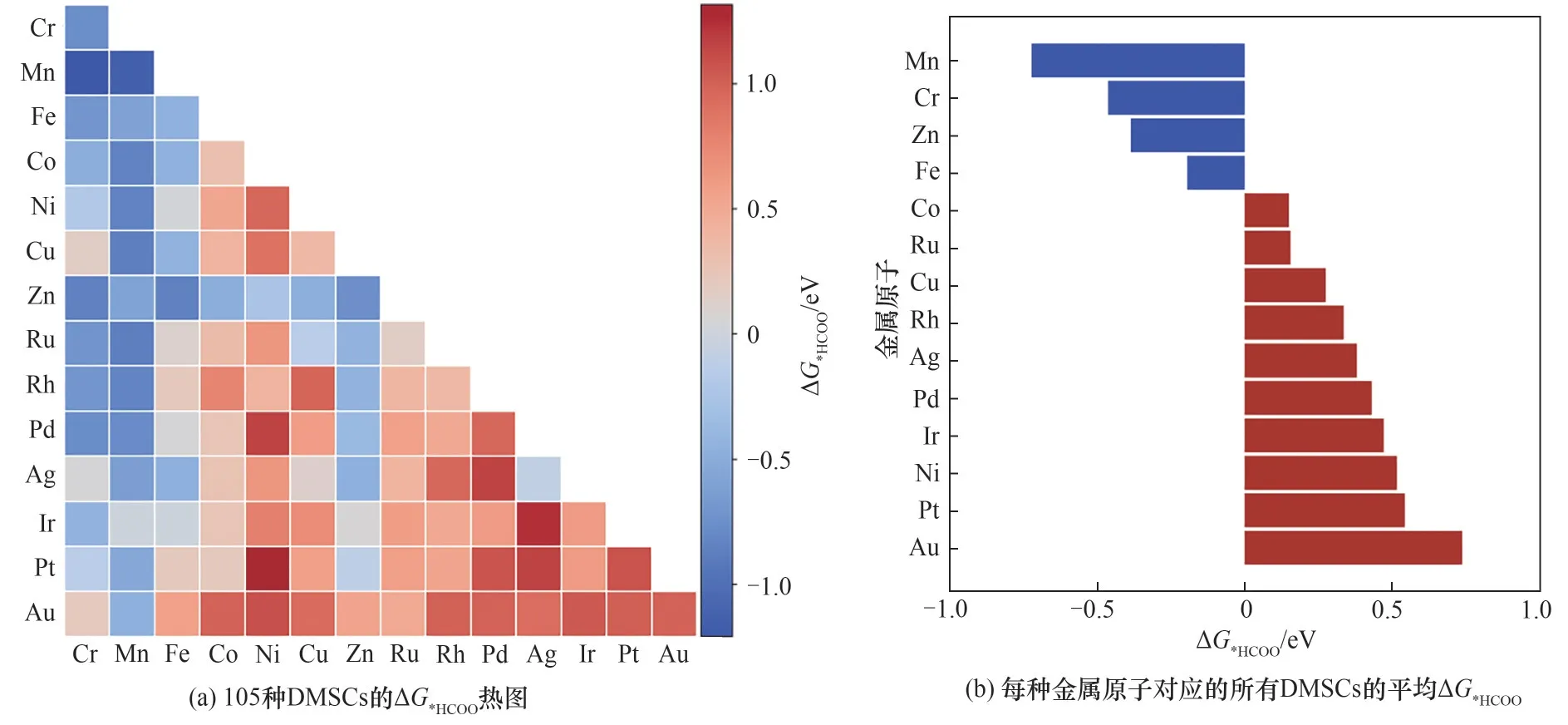
图7 机器学习对ΔG*HCOO的预测结果Fig.7 Predictions ofΔG*HCOO by ML

图48 种算法对ΔG*HCOO预测结果的对比Fig.4 Comparisons between8ML algorithms for predictions ofΔG*HCOO
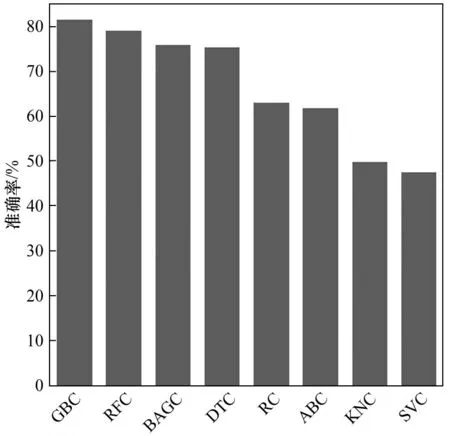
图58 种算法预测*HCOO吸附构型的准确率Fig.5 Accuracy of*HCOO configuration predictions by8ML algorithms
相应地,图8为105种DMSCs表面ΔG*H的预测结果,预测的ΔG*H分布在-0.76 ~0.43 eV之间。与ΔG*HCOO的分布不同,大多数DMSCs的ΔG*H都小于0,如图8(a)中的蓝色和灰色所示。图8(b)显示了各种金属原子对应的所有DMSCs的平均ΔG*H都小于-0.15 eV。其中,Pd原子对应的平均ΔG*H最小,接近-0.6 eV,表明大多数由Pd原子组成的DMSCs对*H具有较强的吸附。

图8 机器学习对ΔG*H的预测结果Fig.8 Predictions ofΔG*H by ML
为了更加直观地比较DMSCs表面的CO2ER和HER,本文将ΔG*HCOO减去ΔG*H绘制出图9,所得结果分布在-1.03 ~1.80 eV之间。ΔG*HCOO-ΔG*H越小,代表该DMSCs表面吸附*HCOO比*H越稳定,有利于二氧化碳还原制甲酸盐。相反,若ΔG*HCOO-ΔG*H越大,则表明吸附*H越稳定,有利于HER。预测结果显示,在105种DMSCs中,共有29种DMSCs的表面更有利于二氧化碳还原制甲酸盐(ΔG*HCOO-ΔG*H小于0),如图9(a)中的蓝色部分。其中,Cr-Mn组合的ΔG*HCOO和ΔG*H相差最大,ΔG*HCOO-ΔG*H为-1.03 eV,ΔG*HCOO远远小于ΔG*H,展现出较好的*HCOO吸附性能。图9(b)显示,由Mn原子或Cr原子组成的所有DMSCs的平均ΔG*HCOO-ΔG*H小于0,表明大部分包含这两种金属原子的DMSCs有利于生成*HCOO,是潜在的转化二氧化碳为甲酸盐的高性能材料。而含有第五、第六周期金属原子的DMSCs,如Au、Pt、Pd、Ir等,ΔG*HCOO-ΔG*H明显高于0,表明*HCOO中间体在其表面十分不稳定,不利于反应朝二氧化碳还原方向进行。
本文运用了类似的方法对*HCOO吸附构型进行了机器学习的预测,结果如图10所示。深蓝色区域代表M1-*HCOO-M2双配位型,蓝色区域代表M1-*HCOO单配位型,淡蓝色区域代表M2-*HCOO单配位型,橙色区域代表M1-*HCOO单配位扭曲型,红色区域代表M2-*HCOO单配位扭曲型。105种DMSCs中,共有52种组合采取M1-*HCOOM2双配位吸附构型,28种组合采取M-*HCOO单配位吸附构型,其余25种组合采取M-*HCOO单配位扭曲型吸附结构。如图10所示,*HCOO在DMSCs表面的吸附构型与金属原子的原子半径有着显著联系,这与原子半径之和(r1+r2)在预测*HCOO吸附构型时的重要度较高(占比为0.21)是一致的。若DMSCs由第四周期具有较小原子半径的过渡金属元素组成,*HCOO的吸附便会采取M1-*HCOO-M2双配位构型。结合图9(a),这些DMSCs大部分具有更小的ΔG*HCOO,展现出较强的*HCOO吸附。然而,具有更大原子半径的第五和第六周期过渡金属元素组成的DMSCs,大部分采用单配位或单配位扭曲型吸附构型,所预测的ΔG*HCOO也往往大于0,不利于生成*HCOO。结合105种DMSCs表面ΔG*HCOO-ΔG*H的预测结果,还发现之前筛选出的29种有利于二氧化碳还原制甲酸盐的DMSCs大多采取了双配位吸附构型,少量采取了单配位吸附构型;而所有采取单配位扭曲型吸附结构的25种DMSCs的ΔG*HCOOΔG*H预测值均大于0.5 eV,不利于生成*HCOO。这一结果表明,DMSCs表面*HCOO的稳定性与其吸附构型密切相关。
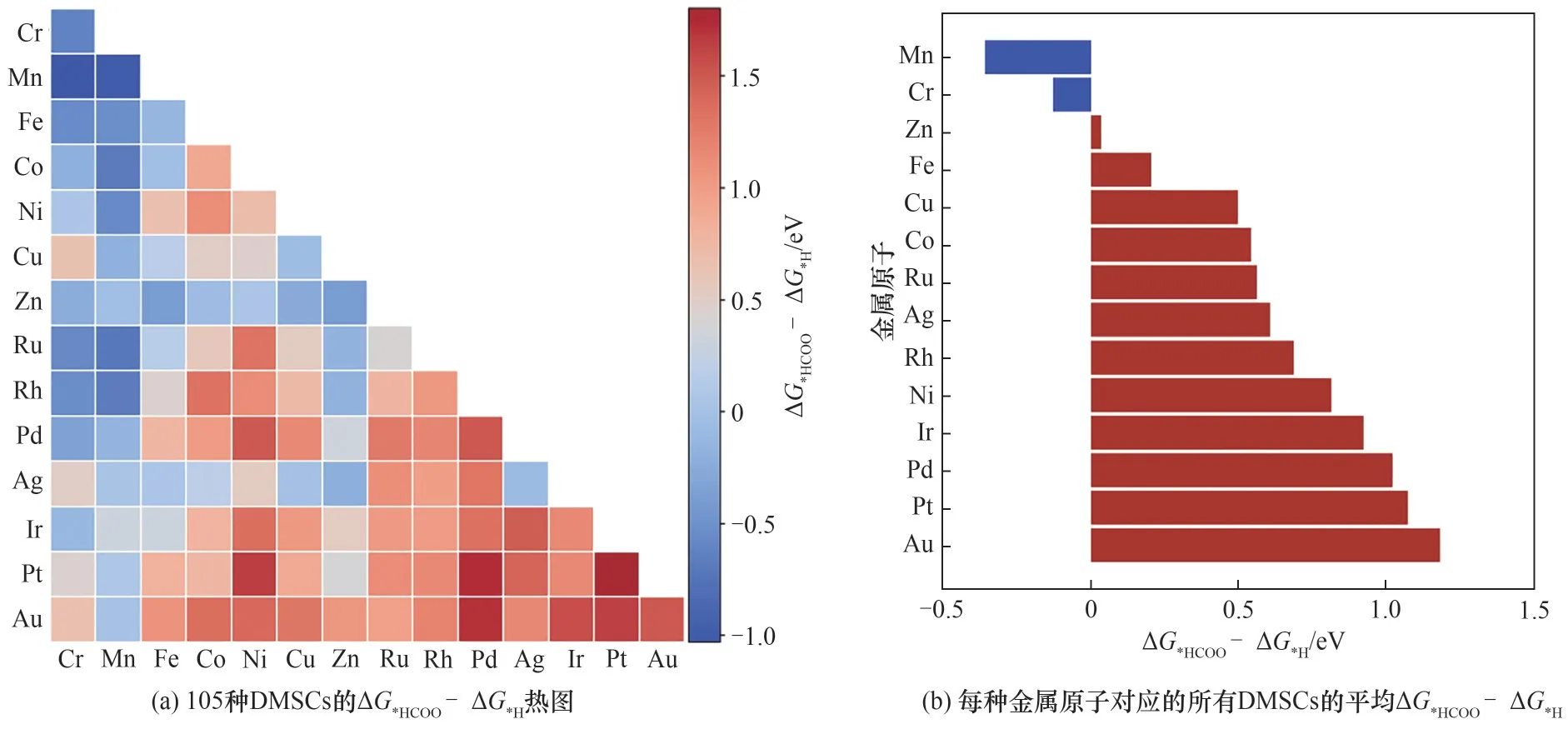
图9 机器学习对ΔG*HCOO-ΔG*H的预测结果Fig.9 Predictions ofΔG*HCOO-ΔG*H by ML
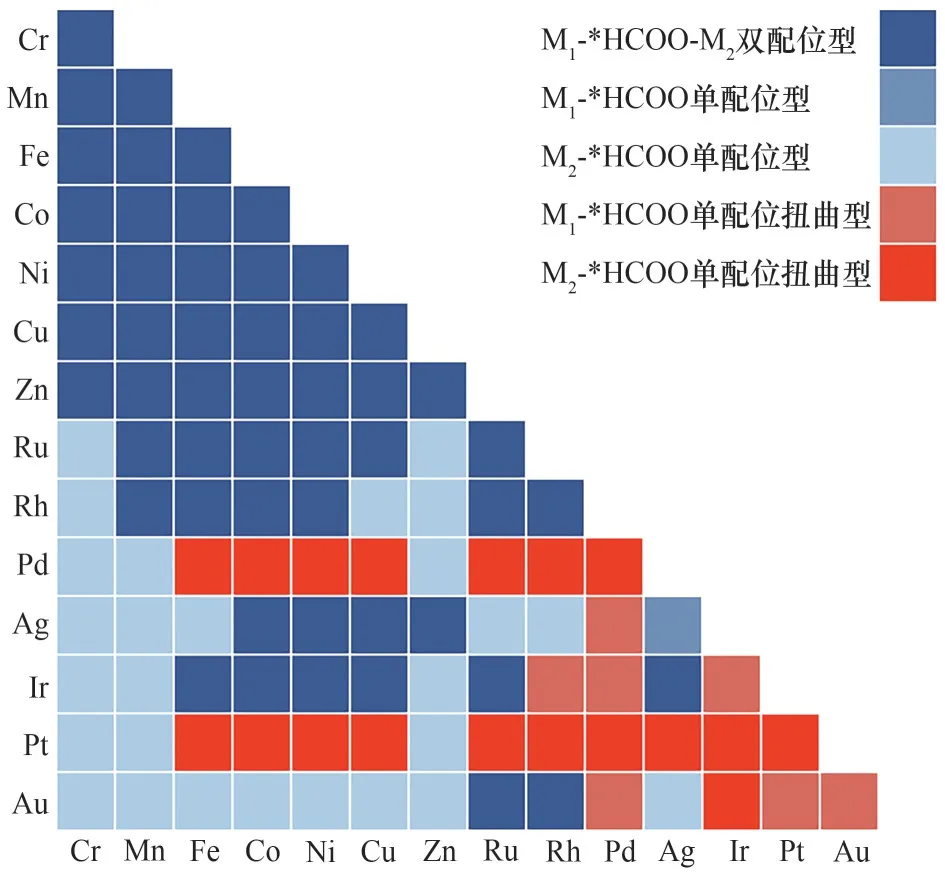
图10 机器学习对*HCOO吸附构型的预测结果Fig.10 Predictions of*HCOO configurations by ML
3 结 论
综上所述,本工作通过结合DFT计算和机器学习,系统地研究了由14种过渡金属元素两两组成的DMSCs催化二氧化碳电化学还原的表现。在这项工作中,利用DFT计算获得随机抽出的23种DMSCs表面二氧化碳还原得到甲酸中间体及其主要竞争反应氢析出反应中间体的Gibbs自由能变,并结合相应构成DMSCs金属原子的物性参数作为机器学习研究的训练集,开展有效的模型训练。其中,金属原子的电子性质,包括平均原子序数、平均价电子数、平均电负性、平均第一电离能、平均电子亲和能、平均d电子数和平均功函数是影响DMSCs表面二氧化碳还原中间体稳定性的主要因素;而平均价电子数、平均第一电离能、平均d电子数和平均范德华半径则是影响氢析出反应中间体稳定性的主要因素。基于这些主要因素作为机器学习的特征,预测了剩余DMSCs的ΔG*HCOO、ΔG*H和*HCOO吸附构型,并确定了105种DMSCs中的29种更有利于二氧化碳还原制甲酸盐,占比达27.6%。其中,大多数由Cr、Mn金属原子组成的DMSCs均具有较小的ΔG*HCOO和较大的ΔG*H,是潜在的转化二氧化碳为甲酸盐的高性能催化材料。机器学习对*HCOO吸附构型的预测结果表明,若DMSCs由第四周期具有较小原子半径的过渡金属元素组成,*HCOO的吸附便会采取M1-*HCOO-M2双配位构型;而具有更大原子半径的第五和第六周期过渡金属元素组成的DMSCs,大部分采用单配位或单配位扭曲型吸附构型。结合ΔG*HCOO和ΔG*H的预测值,在前述29种有利于二氧化碳还原制甲酸盐的DMSCs表面上,甲酸中间体大多采取了双配位吸附构型,少量采取了单配位吸附构型;而所有采取单配位扭曲型吸附结构的DMSCs均不利于生成甲酸中间体。未来,可以基于本文给出预测结果,采用实验手段探索包含Cr、Mn等元素的DMSCs催化CO2ER制甲酸盐的实际性能,努力构筑高效的催化体系,实现二氧化碳制甲酸盐工艺的商业化发展。同时,本文所使用的结合密度泛函理论计算和机器学习的研究策略同样可以用于二氧化碳还原反应其他产物的选择性催化材料以及其他催化体系的预测与筛选。使用这个策略,可以加快对高性能催化材料的筛选过程,进一步缩短研发周期,降低研发成本,为未来设计和研究新型材料提供新的思路。
猜你喜欢
军民两用技术与产品(2022年1期)2022-06-01
核化学与放射化学(2022年2期)2022-04-28
中学生数理化(高中版.高考理化)(2021年12期)2021-03-08
饲料博览(2020年7期)2020-08-18
人工晶体学报(2019年5期)2019-06-18
当代陕西(2019年6期)2019-04-17
分析化学(2018年2期)2018-03-02
北京航空航天大学学报(2017年10期)2017-04-20
外语学刊(2014年3期)2014-12-03
中小企业管理与科技·中旬刊(2014年7期)2014-09-24
