
NH3 在TaC(0001)表面吸附和解离的第一性原理研究*
2022-01-19 04:44李小林袁坤何嘉乐刘洪峰张建波周阳
物理学报 2022年1期
李小林 袁坤 何嘉乐 刘洪峰 张建波 周阳†
1)(江西理工大学,材料冶金化学学部,赣州 341000)
2)(江西理工大学,工程研究院,赣州 341000)
采用自旋极化密度泛函理论(DFT)并结合周期平板模型的方法,研究了NH3 在TaC 表面的吸附和分解反应机理.表面能计算结果显示,以Ta 为终止的TaC(0001)面为最稳定的表面;NH3 分子通过其孤对电子优先吸附在顶位top 位,而NH2 和H 最稳定吸附位置为三重hcp 位,NH 和N 吸附在三重fcc 位.过渡态结果表明氮原子的复合反应脱附为整个反应的限速步骤.电子结构计算结果表明,NH3 分子及其片段通过其N 原子的2pz 轨道与底物Ta 的5 d z2 轨道混合吸附于表面.随着脱氢反应的进行,电荷转移现象变得逐渐明显,吸附质和底物之间的电荷转移在加速NH3 脱氢催化过程中发挥重要作用.
1 引言
氨气(NH3)催化分解制氢技术是指在固体催化剂的作用下将NH3转化为清洁燃料氢气.这项技术不仅可以消除氨气的大气污染,而且提供了一种解决质子交换膜燃料电池电极中毒的有效途径.鉴于氨气分解制氢在燃料电池领域潜在的应用价值,近年来氨气在金属[1]、合金[2,3]和金属氮化物[4]等表面吸附和催化分解引起人们广泛关注.其中金属Ru 和Ir 对氨气脱氢分解反应具有最高的催化活性,但是由于贵金属成本高、资源稀缺,难以大规模开发利用.因此,开发廉价且高效的氨分解催化剂具有重要的科学意义和实际应用价值.
过渡金属碳化物具有和Pt 相似的d 带电子密度态,作为一种潜在的能够替代贵金属氨分解催化材料而被广泛研究.目前文献报道了NH3在WC[5,6]、VC[7]和MoC[8]等催化剂表面吸附分解的实验研究,然而关于氨气在金属碳化物表面上吸附与分解的理论研究还未有报道.在相关文献查询中发现具有多孔结构的TaC 对氨分解反应的催化活性远远超过商业Pt/C 催化剂[9,10],然而NH3在TaC 表面上吸附的电子结构本质和催化分解机理尚不清楚.本文采用密度泛函理论(DFT)方法研究NH3在TaC(0001)面上的吸附位点和几何结构,并从电子结构层次上对吸附成因进行分析,计算脱氢分解各基元反应的过渡态与活化能,并找出脱氢反应的电子结构本质,为进一步开发高性能的氨气分解TaC 催化剂提供理论基础.
2 计算模型和方法
本文所有计算均采用基于密度泛函理论的商业VASP 软件进行.采用了具有广义梯度近似(GGA)的Perdew-Burke-Ernzerh(PBE)泛函来描述核与电子之间的相互作用[11].单电子波函数采用平面波基组展开,截断能为400 eV,布里渊区积分采用Monkhorst-Pack 方案划分K点网格,其中K点网格密度取为5×5×1.在结构优化过程中,力和能量的收敛准则分别设定为0.02 eV/ Å(1 Å=0.1 nm)和10—5eV.计算模型采用具有7 个原子层和垂直于平板15 Å真空层的(3×3)超晶胞模型模拟TaC(0001)表面,其中按照表面层原子种类可分为以碳为终止(C-TaC)和以Ta为终止(Ta-TaC)的结构模型.在构建优化过程中底部5 个原子层的原子坐标是固定的,而顶层表面原子随吸附物分子共同驰豫.
首先计算体相TaC 和气相NH3分子的性质,计算得到六角晶系结构TaC 的晶格参数值为3.12 Å和2.74 Å.对于NH3分子,在尺寸为15 Å×15 Å×15 Å的大单元格中计算得到的键长r(N—H)=1.021 Å,键角θ(H—N—H)=106.6°.这与文献报道的实验值1.017[12],107.8[12]基本一致.
本文吸附能的计算公式为

其中Eall,Esurface和Eadsorbate分别代表吸附体系的总能量、清洁表面能量和吸附分子的能量.如果吸附能为正值,那么吸附为吸热过程,吸附不是自发进行的;反之吸附过程为放热,反应可以自发进行,且吸附能的绝对值越大,吸附体系越稳定,吸附越容易发生.本论文中所有能量进行了零点自由能矫正.
为了绘制最小能量路径并定位过渡态,使用CI-NEB 和dimer 相结合方法寻找化学反应的过渡态.每步脱氢反应的活化能和反应热分别为
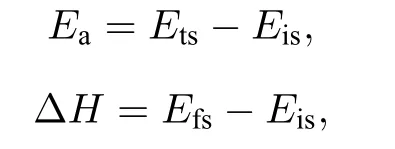
其中Eis,Ets和Efs分别表示反应物、过渡态和产物的总能量.
3 结果与讨论
3.1 表面能的计算
根据非化学计量比表面的表面自由能定义,计算TaC 表面的表面能[13]

其中A表示TaC 模型的表面积;ESlab表示板层模型优化后的能量值;NTa,NC分别表示模型中钽原子和碳原子个数;µTaC(bulk)表示体相TaC 的化学势;µC表示碳的表面相化学势.图1 展示了Ta-TaC 和C-TaC 的表面能与化学势之间关系.从图1可以看出,在整个能量范围内Ta-TaC 表面能始终低于C-TaC 表面能,这说明以钽为终止的TaC(0001)表面更稳定.
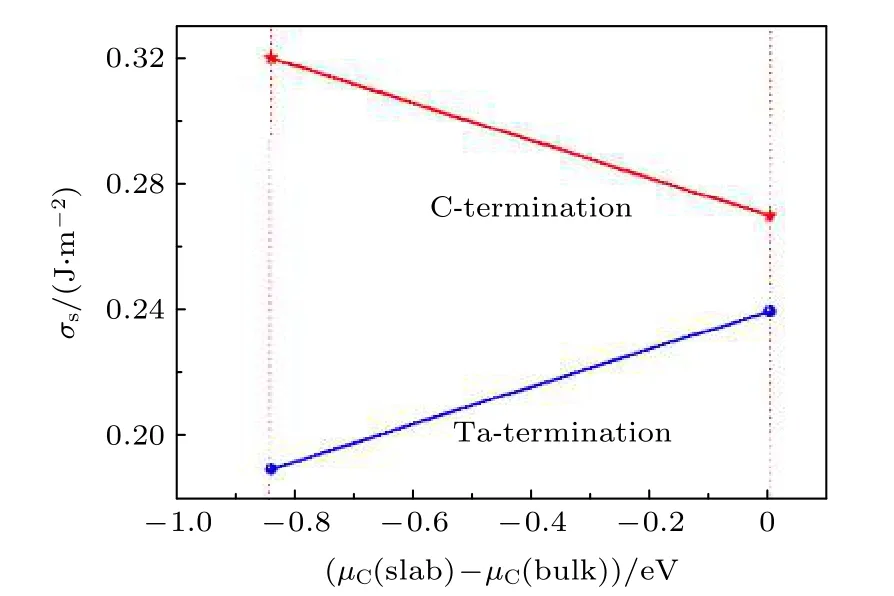
图1 TaC(0001)的表面能(σs)与化学势之间的关系Fig.1.Relationships between surface energies of two TaC(0001)surfaces and chemical potentials.
3.2 NHx 在Ta-TaC 表面的吸附
在上述TaC(0001)表面能及稳定性研究基础上,选择以金属钽为终止的TaC(0001)表面作为吸附能和过渡态的平面模型,同时考察了NHx及其他小分子在顶位(top)、三重空位(fcc 和hcp)和桥位(bridge)的吸附构型(图2).表1 列出了优先吸附位置的吸附能和关键几何参数.
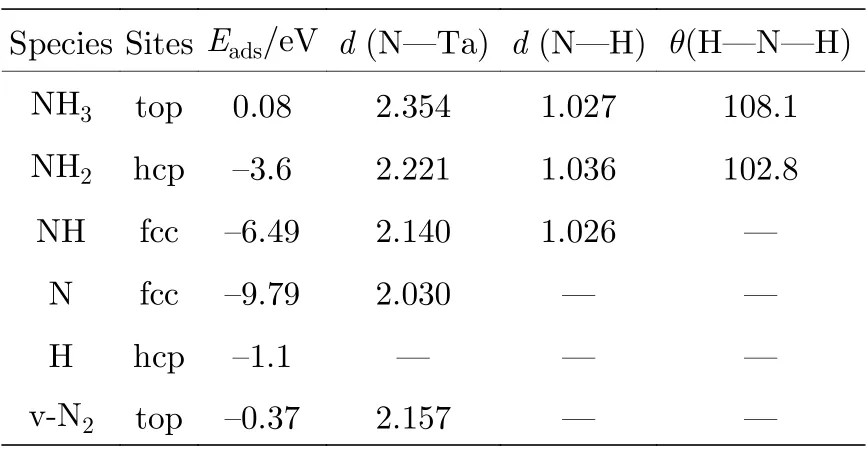
表1 Ta-TaC 表面的吸附位点、吸附能和关键几何参数Table 1.Adsorption site,adsorption energy and key geometric parameters of Ta-TaC surface.
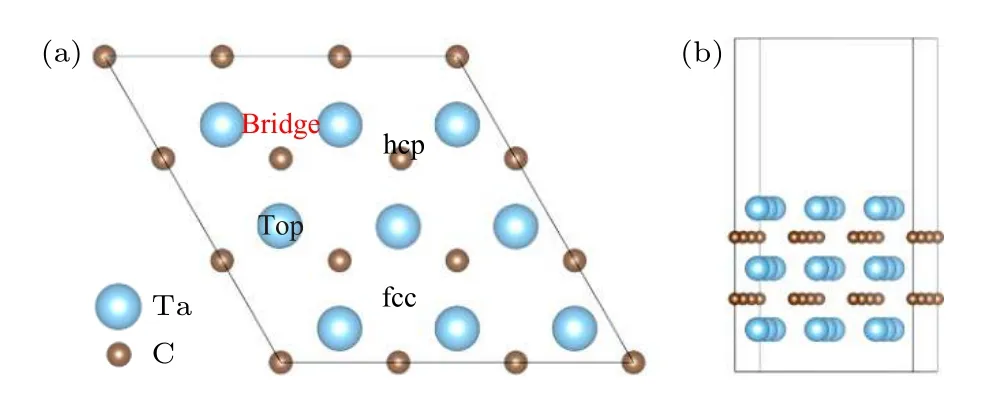
图2 Ta-TaC 表面的俯/侧视图及吸附位点Fig.2.Side view of Ta-TaC and adsorption site.
对于NH3来说,由于其具有Cs 点群对称性,需要考虑两种构型来优化吸附几何结构:一种结构是一个氢原子在氮原子的左边,两个氢原子在氮原子右边(图3(a));另外一种结构是两个氢原子在氮原子的左边,一个氢原子在氮原子的右边(图3(b)).除NH2具有C2V对称外,其他所有组成成分都具有C4V 对称.同时还考虑了平行(图3(c),p-N2)和垂直(图3(d),v-N2)两种吸附构型.
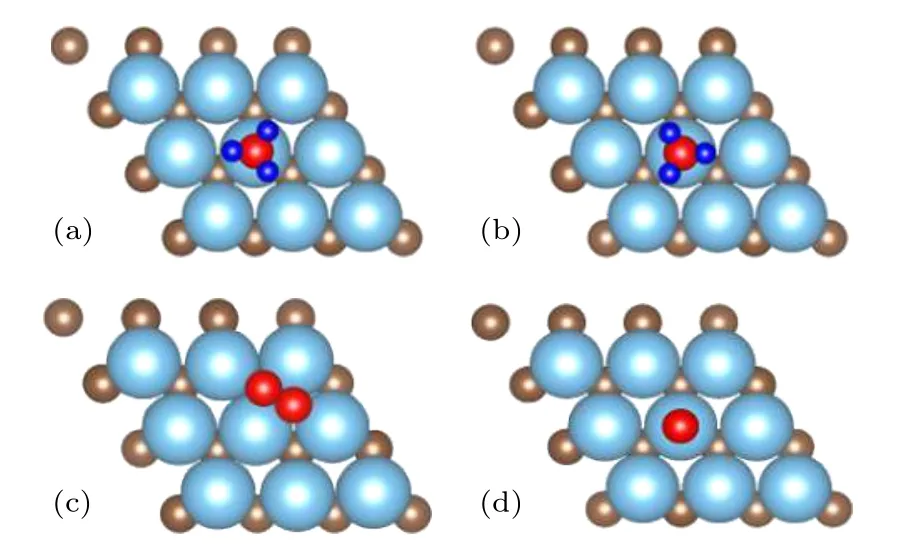
图3 (a)(b)NH3 和 (c)(d)N2 的两种吸附构型Fig.3.Two adsorption configurations of (a)(b)NH3 and(c)(d)N2.
对于NH3在Ta-TaC 表面的吸附,对4 个位置(top,hcp,fcc 和bridge)进行优化.其中top 位最稳定,NH3分子吸附在top 位,然后把这个结构模型作为NH3分解催化剂的始态.NH3在Ta-TaC 表面逐步脱氢过程中,中间产物在该表面的最稳定吸附位点、吸附能及一些关键几何参数如表1 所示.NH3优先吸附在顶部位置,N 原子与Ta 成键,H 原子指向外.任何试图在其他对称位点找到能量最小值的尝试都将在完全优化后得到顶部位置,这与文献[14,15]报道的NH3在过渡金属上的吸附一致.NH3,NH2,NH 和N 的吸附能分别是0.08 eV,3.6 eV,6.49 eV 和9.79 eV.在优先顶部几何结构中,N 原子距离表面2.354 Å.N—H 键长和N—H—N 角分别为1.027 Å和108.1°,与气相NH3分子的数值相接近.这说明NH3分子的结构在吸附后没有明显改变,NH3-底物之间存在较弱的相互作用.对于NH2,吸附位置在hcp 位置,N—H 键长、H—N—H 键角、N 与Ta 表面的垂直距离分别是为1.036 Å,102.8°,2.221 Å.N—H键长从片段NH2的1.037 Å下降到1.036 Å,而H—N—H 键角由102.5°增大到102.8°.对于NH和N,吸附位置都是在fcc 位置,N 与Ta 表面的垂直距离分别为2.140 Å和2.03 Å.综上所述,随着NHx组分中H 原子数的减少,N 原子与TaC 表面的垂直高度降低,Ta 配位N 原子数增加,NHx中的未成键的孤对电子和底物Ta 原子之间相互成键,导致NHx组分的吸附能逐步增大.
3.3 NH3 在Ta-TaC 表面的逐步脱氢分解机理
众所周知,NH3分子在催化剂表面分解脱氢生成N2,包含以下4 个基元反应过程:[16,17]

对于NH3脱氢和复合反应的初态(IS)、过渡态(TS)和终态(FS)结构如图4 所示,其中NH3在Ta-TaC 表面的第一步解离,选择了NH3分子在平面模型表面的顶位吸附作为初始态(IS1).随着N—H 距离由1.027 Å增大至1.037 Å,形成过渡态TS1.通过相应的过渡态中N—H 的裂解,进一步解离成NH2和H.这一步需要克服0.93 eV的能垒,吸附放热2.1 eV.NH2+H 共吸附构型(IS2)被用作第二阶段分解的初始状态,其中被吸附的NH2和H 分别占据了hcp 位置.NH2通过过渡态TS2 使N—H 键断裂,脱氢分解至NH 和H 物种共吸附在Ta-TaC 表面.第二步需要克服1.02 eV 的能垒,吸附放热1.78 eV.NH 和H 物种的最稳定共吸附态(IS3,fcc)作为第三步脱氢反应的反应物,N—H 键距离逐渐拉伸,当N-H 距离增大到3.116 Å时,TS3 过渡态被确定.第三步脱氢反应的能垒为2.03 eV,放热为0.27 eV.经过以上三步脱氢过程,最终氮原子以最稳定的fcc 位吸附在TaC 表面,两个N 原子再进一步相互靠拢,当N 和N 原子之间间距为1.37 Å时,过渡态形成(TS4).第四步复合反应的能垒为5.32 eV,吸热为2.89 eV.
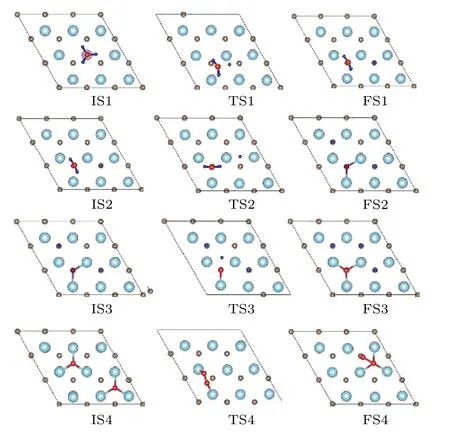
图4 NH3 脱氢和复合反应的初态、过渡态和终态结构Fig.4.Structure of initial state,transition state and final state of NH3 dehydrogenation and recombination reaction.
综上所述,根据图5 和表2 的计算结果,得出以下结论:1)脱氢反应为放热反应,说明氨气分子适合在Ta-TaC 表面吸附分解,其脱氢反应能垒与目前报道贵金属催化剂相接近[18,19];2)有且只有一个虚频,说明搜索过渡态结构的唯一性;3)随着NHx组分中H 原子数的减少,NHx在其表面的吸附能逐渐增大,导致N—H 键的活化解离所需要的活化能也逐渐增大,氮原子的复合反应脱附成为整个反应的限速步骤.
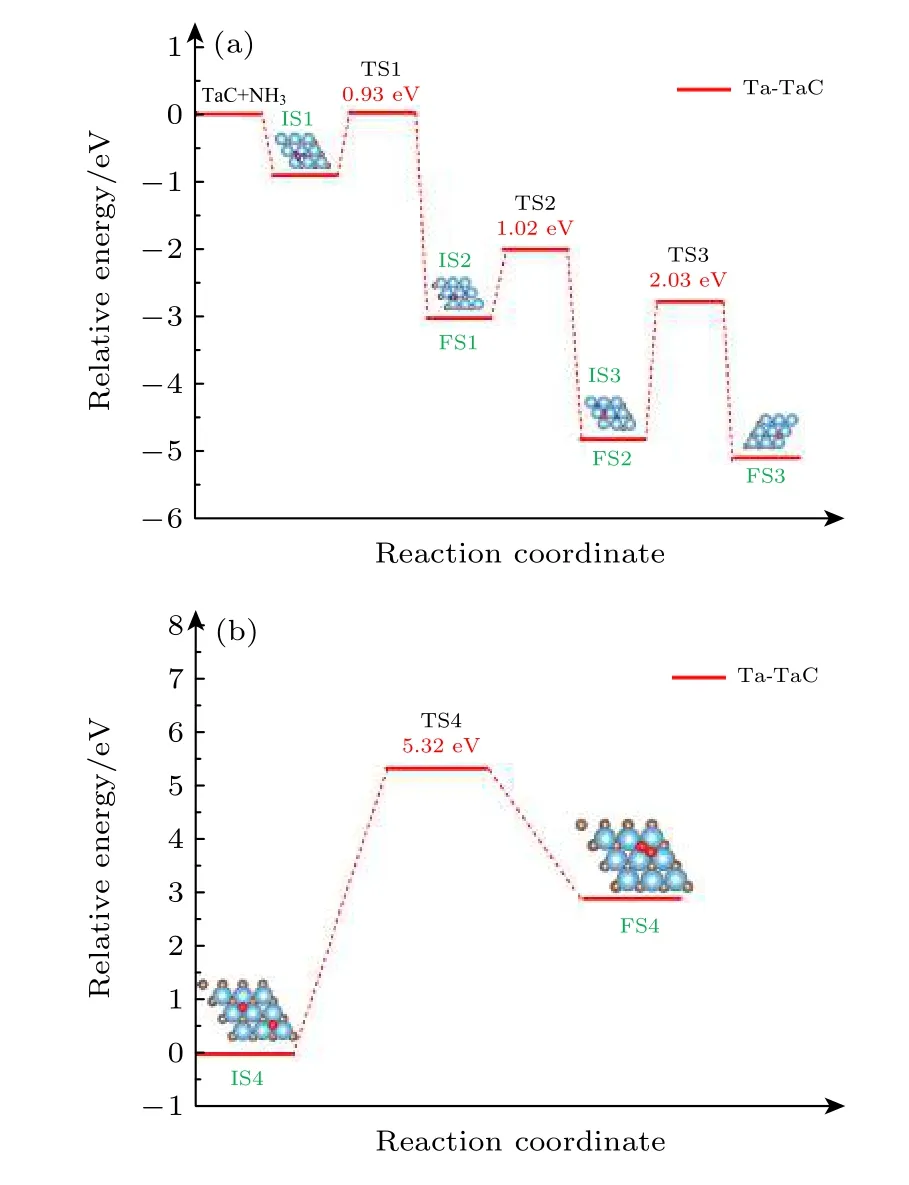
图5 (a)NH3 分解反应和 (b)N-N 复合反应的能量图Fig.5.Calculated potential energy diagram for (a)NH3 dehydrogenation and (b)N-N coupling reaction.
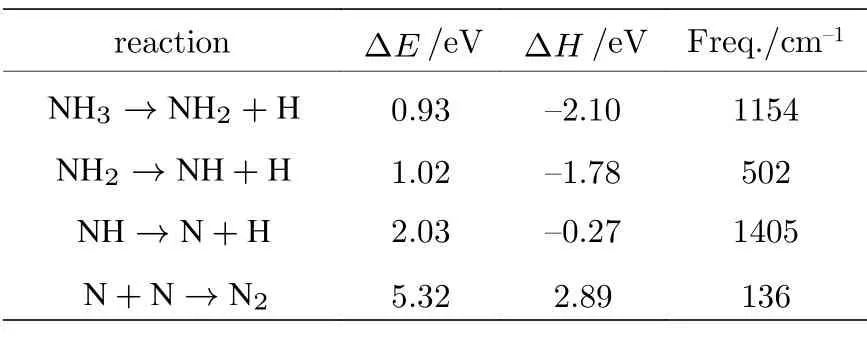
表2 各步基元反应的活化能、虚频和反应热Table 2.The activation energy、imaginary frequency and reaction heat of each step elementary reaction.
3.4 NHx/Ta-TaC 表面电子结构分析
为进一步阐明NH3在Ta-TaC 表面吸附解离的机理,本文从电荷密度分布和电子态密度角度分析最稳定吸附位置的电子结构.如图6 所示,通过吸附N 和H 原子及其最邻近的表层原子截取电荷密度分布图,其中原子周围的黄色区域表示成键区域电荷密度增强,蓝色区域表示成键区域电荷密度减小,轮廓等值面为0.015e/Å3.此外,还使用Bader电荷分析方法计算了吸附体系中各原子电荷.从图6(a)可知,NH3分子和底物表面之间的电荷转移非常小(只有0.01|e|,从Ta 原子到NH3分子).随着第1 次脱氢反应的进行,0.6e 从底物表面转移到NH2片段(图6(b)).在第2 次和第3 次脱氢反应过程中,电荷转移现象变得更加明显,这个值分别增大到1.03e 和1.33e (图6(c)和6(d)).通过以上结果分析可知,在NH3脱氢催化过程中吸附质和底物之间的电荷转移是其吸附能和活化能增加的根本原因.
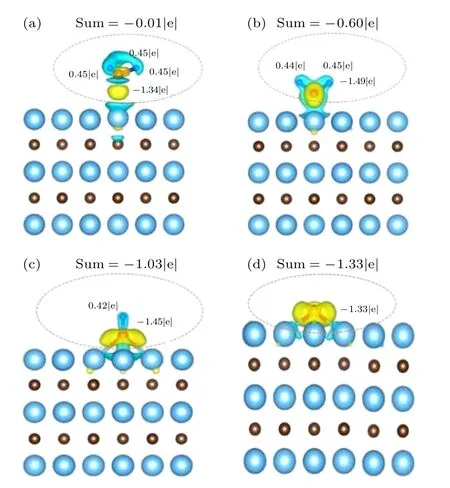
图6 (a)NH3,(b)NH2,(c)NH,(d)N 在Ta-TaC 表面的差分电荷密度示意图和Bader 电荷分析Fig.6.Schematic diagram of differential charge density of(a)NH3,(b)NH2,(c)NH,(d)N on the surface of Ta-TaC and Bader charge analysis.
为了进一步了解NH3及其片段与Ta-TaC 表面相互作用机理,计算了吸附前后体系的态密度.众所周知,NH3在气相中的电子结构可以表示为(σ2a1)2(σ1e)4(n3a1)2,其中2a1轨道主要由N原子的2s 轨道和H 原子的1s 轨道形成,1e 轨道由N 原子的2Px及2Py轨道与H 原子的1s 轨道混合而成,3a1 轨道由N 原子的2pz轨道组成[20,21].从图7(a)可以看到,在能量范围为—20—0 eV 之间出现3 个不同峰,分别代表σ2a1,σ1e和n3a1轨道.从图7(a)—7(d)中可以看出,在吸附前后及脱氢过程中2a1和1e 轨道所对应的峰形貌变化不大,说明2a1 和1e 轨道同表面态之间相互作用弱,对NH3及其片段的吸附贡献小.同气相NH3相比(Fig.7(a)),3a1 轨道所对应的峰出现明显展宽现象,同时所对应的能量向低能方向发生了位移(图7(b)—7(e)),说明在垂直方向上N 原子的2pz轨道和Ta 原子d 轨道之间出现杂化行为,这是NH3及其片段稳定吸附于表面的主要原因,类似于NH3在其他体系吸附的成因[13,22].

图7 (a)吸附前NH3,(b)吸附后NH3,(c)NH2,(d)NH,(e)N 吸附于Ta-TaC 表面态密度分布,虚线代表费米能级Fig.7.Local density of states (LDOS)for NH3 dehydrogenation:(a)NH3 before interaction,(b)NH3 after interaction,(c)NH2,(d)NH,(e)N The dashed line represents the Fermi level.
4 结论
采用第一性原理的方法研究了NH3在Ta-TaC 表面上的吸附和脱氢反应机理.得到了相应的最优吸附位置、能量和几何结构,确定了NH3逐步分解反应的过渡态结构及能垒.主要结论归纳如下:
1)表面能计算结果说明以钽为终止的TaC(0001)表面更稳定.
2)NH3和N2倾向于吸附在top 位上,而NH,N 倾向于吸附在fcc 位上,NH2和H 倾向于hcp 位上.各组分在脱氢过程中的吸附能呈现如下趋势:NH3< NH2< NH < N.
3)在NH3的整个分解反应中,N 与N 原子之间复合脱附反应活化能较高,它决定了整个反应的快慢,是整个反应的速率控制步骤.
4)电子结构分析结果表明,NH3分子及其片段通过其N 原子的2pz轨道与底物Ta 的 5 dz2轨道混合吸附于表面.随着脱氢反应的进行,电荷转移现象变得逐渐明显.
猜你喜欢
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
教育周报·教育论坛(2020年3期)2020-10-21
科技资讯(2018年16期)2018-10-26
科技信息·下旬刊(2018年8期)2018-10-21
科技资讯(2017年12期)2017-06-09
科学时代·上半月(2013年12期)2013-12-26
