
天然免疫信号蛋白在仙台病毒感染过程中聚集于应激颗粒
2022-02-18 03:04孙英杰
中国动物传染病学报 2022年6期
吴 琼,孙英杰,丁 铲,廖 瑛
(中国农业科学院上海兽医研究所,上海 200241)
一些病毒感染真核细胞时,细胞迅速终止蛋白翻译,在细胞质中形成的致密颗粒状聚合体,即应激颗粒(Stress granule, SG)[1]。RNA病毒主要通过复制过程中的中间产物双链RNA,激活PKR,磷酸化eIF2α,使蛋白翻译起始所需的三元复合物eIF2-GTP-Met-tRNAMet不能循环利用,蛋白翻译停滞[2],多聚核糖体解离,mRNA和翻译相关元件在细胞质中与G3BP1(Ras GTPase-activating protein binding protein 1)和TIA1(T cell restricted intracellularantigen-1)等核心蛋白聚集形成SG。
已有报道表明,SG能够招募RNA感受器RIG-I和PKR,启动干扰素信号通路,促进抗病毒天然免疫应答[3]。因此,许多病毒感染进化出抑制SG的机制,抵御其抗病毒作用[4-5]。例如:Theiler氏鼠科脑脊髓炎病毒(Theiler's murine encephalomyelitis virus,TMEV)在感染细胞时,病毒非结构蛋白L抑制SGs的形成[6]。麻疹病毒(Measles virus, MV)在感染细胞后,细胞中不产生SG;与野生型毒株相比,麻疹病毒C蛋白缺失的毒株能诱导SG的形成,说明C蛋白在抑制SG形成的过程中发挥了重要作用,但机制尚不明确[5]。少部分报道显示,病毒利用SG来帮助自身的复制[7-9]。如西尼罗河病毒(West Nile virus,WNV)RNA 3'端茎环结构可以通过对SG成核蛋白TIA1的“劫持”,抑制对亚砷酸钠应激诱导产生的SGs的形成,从而促进病毒的复制[7,10]。SG是一个动态的RNA和蛋白聚合物结构,当细胞压力缓解后,SG解聚,mRNA和翻译相关因子回归到核糖体,重启蛋白翻译;如果压力持续存在,SG的mRNA进入P小体(processing bodies),发生降解,蛋白则由自噬小体降解[11-12]。
宿主受到病毒感染时,会启动一系列的抗病毒反应。在感染初期,宿主细胞主要通过天然免疫应答抵御病毒侵染:即通过细胞内的模式识别受体(pattern recognition receptors, PRRs),识别DNA或RNA病毒的病原相关分子模式(pathogen-associated molecular pattern, PAMP),激活天然免疫应答信号通路,诱导机体产生干扰素(interferon, IFN)、干扰素刺激基因(interferon stimulated gene, ISG)和炎性细胞因子,使周围细胞处于警戒状态,抵御病毒的进一步感染和扩散[13]。识别RNA病毒感染的PRRs主要包括toll样受体(Toll-like receptors, TLRs)、RIG-I样受体(retinoic acid-inducible gene I (RIG-I)-like receptors, RLRs),以及PKR(protein kinase R,PKR)[14-15]。TLR3主要位于内体膜上,识别通过内吞途径进入内含体的病毒RNA[16-17]。RIG-I和MDA5位于细胞质,负责识别进入细胞质的病毒RNA[18]。PKR位于细胞质,主要识别病毒复制过程中产生的双链RNA[19-20]。PRRs识别病毒核酸后,通过招募各自的适配器,将信号传到各个激酶,最终通过磷酸化修饰激活转录因子NF-κB或IRF3/7,进入细胞核,诱导促炎性细胞因子、趋化因子和Ⅰ型IFN的产生。细胞因子分泌到细胞外,结合细胞表面受体,激活JAK-STAT信号通路,诱导干扰素刺激基因(IFN stimulated genes, ISGs)的表达,或激活免疫细胞,抵御病毒的感染[21-22]
仙台病毒(Sendai virus, SeV)于20世纪50年代在日本仙台发现[23],为单链负链RNA包膜病毒,属于副粘病毒家族,平均直径为260 nm[24]。SeV主要感染啮齿类动物和猪,引起呼吸道症状。自从被发现以来,SeV被广泛应用于研究:SeV对人类无致病性,对多株肿瘤细胞中具有感染性,在动物模型中具有溶瘤作用,可开发为溶瘤病毒[25-26];SeV介导真核细胞发生膜融合,可应用于制备单克隆抗体的杂交瘤细胞[27];完整的活性SeV常作为IFN表达的诱导因子,用于IFN的大规模生产[28-29];SeV也可作为表达载体,应用于制备全能干细胞,或者应用于制备疫苗[30-31]。
我们的前期研究发现,不少病毒感染细胞时,天然免疫信号蛋白聚集到SG。已有的研究报道表明,SeV通过C蛋白限制双链RNA的形成,避免激活PKR-eIF2α-SG通路[32];C蛋白缺陷型SeV能诱导类似于SG的颗粒聚集[33];没有进入核衣壳的病毒RNA,被招募到了SG[33]。SeV为何通过C蛋白抑制SG的形成?病毒RNA聚集到C蛋白缺陷型SeV诱导的SG,发挥了什么作用?是否参与促进IFN反应?本研究通过检测PRRs和天然免疫信号传递蛋白在SeV感染过程中的亚细胞定位,解析以上科学问题。我们发现SeV感染能够诱导一部分细胞形成SGs。对这部分形成SG的细胞进行间接免疫荧光观察,发现PRRs和天然免疫信号传递信号蛋白聚集到SG。我们推测SeV感染细胞时,PRRs迁移到SGs,识别病毒RNA,进一步招募天然免疫信号传递蛋白,最终促进干扰素反应。由于SG发挥了感染信号识别和信号传递平台的作用,不利于病毒复制,病毒进化出拮抗机制,通过C蛋白减少双链RNA的累积,逃逸对PKR的激活和SG的形成。
1 材料与方法
1.1 主要试剂鼠G3BP1单克隆抗体(mouse monoclonal anti-G3BP1, ab56574)和兔G3BP1单克隆抗体(Rabbit Anti-G3BP antibody, ab181150)购自Abcam公司;兔MDA5单克隆抗体(rabbit monoclonal anti-MDA5, 5321T)和RIG-I单克隆抗体(rabbit monoclonal anti-RIG-I, 3743S)、兔TBK1单克隆抗体(rabbit monoclonal anti-TBK1, 38066S)、兔IKKε单克隆抗体(rabbit monoclonal anti-IKKε,9966T)、兔TAK1单克隆抗体(rabbit monoclonal anti-TAK1, 5206S)、兔DDX3单克隆抗体(rabbit monoclonal anti-DDX3, 8192S)、兔IKKβ单克隆抗体(rabbit monoclonal anti-IKKβ, 9966T)、兔A20单克隆抗体(rabbit monoclonal anti-A20, 5630S)和兔CYLD单克隆抗体(rabbit monoclonal anti-CYLD,8462S)购自Cell Signaling Technology公司。
1.2 细胞与病毒HeLa细胞购自ATCC,并由本实验室保存。用含10%FBS(Hyclone)和1%青霉素/链霉素的DMEM(Gibco),置于含5%CO2、37℃恒温细胞培养箱中培养。SeV由扬州大学张泉馈赠,并由本实验室保存。
1.3 SeV感染用镊子取25 mm细胞飞片在酒精灯外焰灭菌,凉却后轻轻放入6孔细胞板中。将HeLa细胞按5~6×105个铺至细胞板中,摇晃均匀,置于5%CO2、37℃细胞培养箱中培养。待细胞密度长至70%~80%,进行接毒实验。取无菌15 mL离心管,用无血清的DMEM稀释病毒,每孔细胞加入1 mL的无血清DMEM稀释的病毒液,病毒感染量为1 MOI(multiplicity of infection)。置于5%CO2、37℃细胞培养箱孵育1 h,PBS清洗3遍细胞,加入含2%FBS的DMEM,置于细胞培养箱继续培养。待感染后16 h,收取细胞样品,进行间接免疫荧光实验。
1.4 polyI:C转染用镊子取25 mm细胞飞片在酒精灯外焰灭菌,凉却后轻轻放入6孔细胞板中。消化HeLa细胞或Vero细胞,将细胞按5~6×105个铺至细胞板中,待细胞长至70%左右时进行转染。转染时需要准备无RNA酶EP管及枪头,EP管中加入Opti-MEMTM减血清培养基100 μL,按照质量体积比1∶2先加入转染试剂Fugene,吹打混匀后静止5 min,然后再加入2 μg polyI:C,再次轻轻吹打混匀后室温静置15 min,最后将其滴加在事先铺满Opti-MEM™的细胞孔内。
1.5 间接免疫荧光(indirect immunofluorescence assay, IFA)病毒感染后的16 h,吸去培养基,用PBS清洗3遍,每孔加入1 mL含4%多聚甲醛的PBS固定液,室温固定10 min。吸去多聚甲醛固定液,PBS清洗3遍,每孔加入1 mL含0.5%Triton X-100的PBS透化液,室温透化10 min。吸去透化液,用PBS清洗3遍,每孔加入1 mL含3%BSA的PBS封闭液,放入37℃恒温箱中封闭30 min。吸去封闭液,用PBS清洗3遍,跟G3BP1抗体稀释液37℃共孵育1 h;TBST清洗3遍,加入荧光二抗稀释液37℃共孵育30 min。TBST清洗3遍,依次孵育第二组一抗和荧光二抗。全部抗体孵育完毕后,TBST清洗3次,跟DAPI稀释液37℃共孵育15 min。弃去DAPI稀释液,TBST清洗3次,加抗荧光淬灭封片剂,将细胞飞片取出,吸干多余水分放到载玻片上,置于黑暗处过夜晾干。通过ZEISS激光共聚焦显微镜观察SG的形成和相关蛋白的亚细胞定位。
2 结果
2.1 MDA5和RIG-I在SeV的感染过程中聚集于SGs在病毒的感染过程中,PRRs识别病毒感染产生的PAMP,发生信号传递,最终发生抗病毒反应。我们推测参与识别病毒感染的PRRs,在病毒感染过程中会发生细胞内迁移,在空间上接近PAMP。RIG-I和MDA5主要识别细胞质中的病毒双链RNA,激活位于线粒体的MAVS,传递信号到TBK1和IKKε,最终激活IRF3,诱导IFN的表达[17]。在本研究中,我们首先检测SeV感染是否诱导细胞形成SG;同时检测在SeV感染过程中,RIG-I和MDA5 的亚细胞定位是否发生变化。以1 MOI SeV感染宫颈癌细胞HeLa,设置模拟感染(mock infection)为对照组。感染后16 h,利用G3BP1鼠抗和MDA5兔抗,或G3BP1鼠抗和RIG-I兔抗,对细胞进行免疫荧光染色,检测SG核心蛋白G3BP1和MDA5或RIG-I的亚细胞定位。G3BP1的聚集,是SG形成的标志。如图1所示,在模拟感染的对照组,G3BP1均匀分布于细胞质,没有观察到G3BP1聚集颗粒的形成;MDA5和RIG-I均匀分布于细胞质和细胞核;在SeV感染的实验组,大部分感染细胞中的G3BP1均匀分布在细胞质,MDA5和RIG-I也均匀分布于细胞质和细胞核;只有少部分感染细胞的G3BP1发生点状聚集,形成SG;同时,MDA5和RIG-I也发生点状聚集,跟G3BP1颗粒共定位。以上结果说明SeV感染在大部分细胞不诱导形成SGs,仅诱导一小部分细胞形成SG; MDA5和RIG-I迁移到SGs。
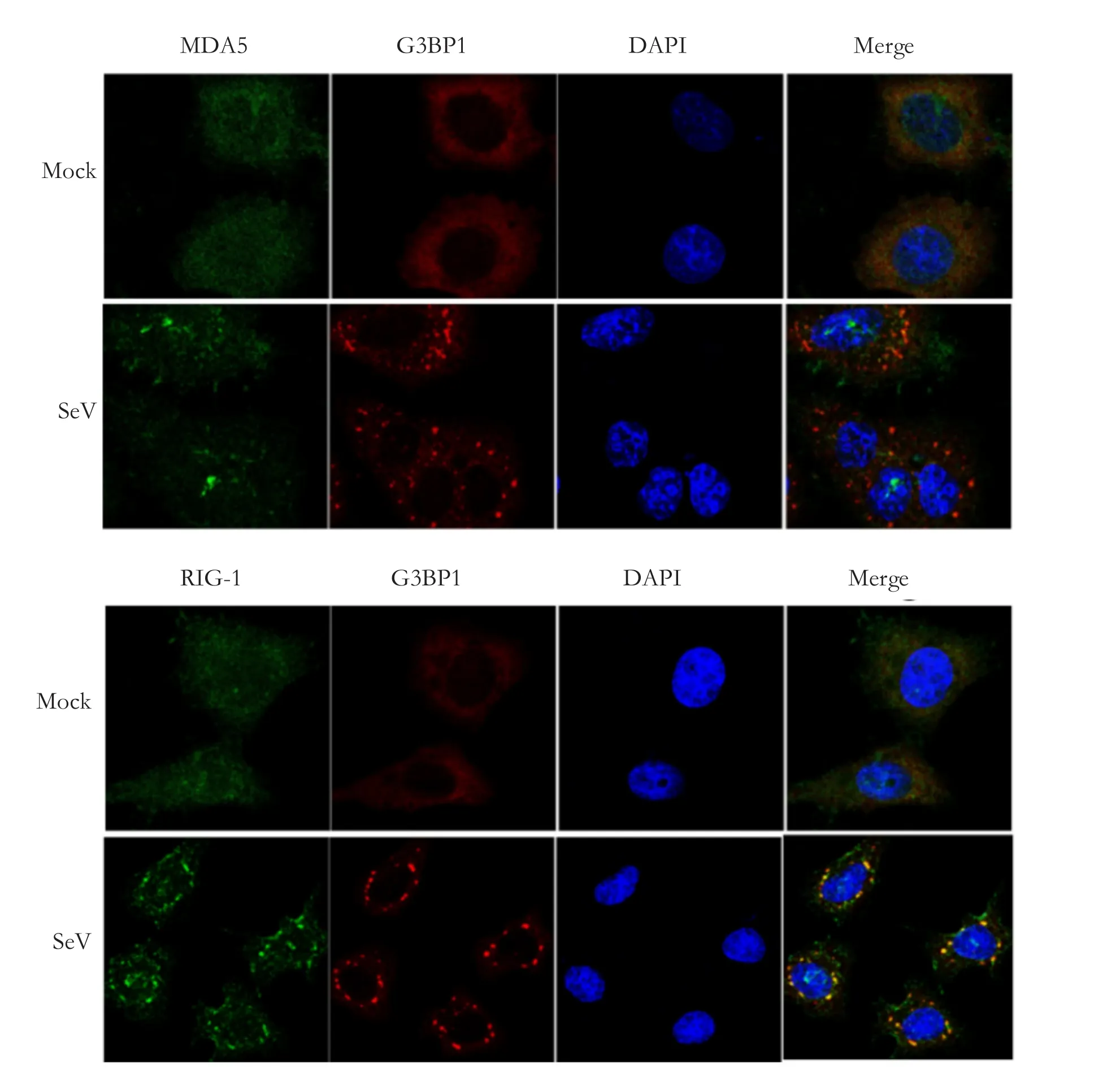
图1 RIG-I和MDA5的免疫荧光共聚焦实验Fig.1 Confocal immunofluorescence assay of RIG-I and MDA5
2.2 IRF3信号通路蛋白激酶TBK1和IKKε在SeV的感染过程中聚集于SG已有报道显示C蛋白缺陷型SeV感染后,诱导大量SG形成,没有核衣壳蛋白包裹的病毒RNA进入SG[33]。本研究发现RIG-I和MDA5迁移到SG,说明这两个PRRs很有可能在SG对病毒RNA进行识别,并进一步激活MAVS,在线粒体膜上发生聚集,招募并激活下游信号蛋白TRAF3,形成IKKε/TBK1复合体,磷酸化激活IRF3进入细胞核,诱导IFN的表达和分泌[17]。RIG-I和MDA5对MAVSIKKε/TBK1的激活,需要有空间上的接触。我们接下来检测IKKε和TBK1在SeV感染过程中的亚细胞定位。以1 MOI SeV感染HeLa细胞,设置模拟感染为对照组。感染后16 h,利用G3BP1鼠抗和TBK1兔抗,或G3BP1鼠抗和IKKε兔抗进行免疫荧光实验,检测SG核心蛋白G3BP1和IRF3信号通路的激酶TBK1或IKKε的亚细胞定位。如图2所示,在模拟感染的细胞中,G3BP1均匀分布于细胞质,没有观察到SG的形成;TBK1或IKKε也均匀分布于细胞质和细胞核;在SeV感染的细胞中,一部分细胞的G3BP1发生点状聚集,形成SG;TBK1和IKKε发生点状聚集,跟G3BP1颗粒共定位。以上结果说明TBK1和IKKε聚集于SeV诱导的SG,参与信号传递。

图2 TBK1和IKKε的免疫荧光共聚焦实验Fig.2 Confocal immunofluorescence assay of TBK1和IKKε
2.3 NF-κB信号通路蛋白激酶TAK1,IKKβ和RNA解旋酶DDX3在SeV的感染过程中聚集于SG作为TLRs模式识别受体的信号蛋白,TLR3主要识别RNA病毒,招募信号蛋白MyD88,激活TRAF6;TRAF6经过k63连接的聚泛素化,作为一个支架蛋白招募TAK1;磷酸化的TAK1随后磷酸化激活IKKα/β/γ复合物[16];IKKβ进一步对IκB进行磷酸化;IκB随后发生泛素化降解,释放转录因子NF-κB进入细胞核,启动IFNβ、TNFα和IL6等细胞因子的表达[17]。DDX3(DEAD-box helicase 3)是RNA解旋酶,参与天然免疫信号转导,常常被病毒劫持以促进复制[34-35]。已有的研究表明,过表达DDX3促进SG组装,而敲减DDX3的表达则抑制SG组装;同时DDX3的解旋酶活性对SG组装必不可少,且DDX3与eIF4E结合是SG组装所必需的[36]。DDX3的RNA解旋酶活性使mRNA从RNA颗粒中逃逸并在多聚体处恢复翻译[37]。TAK1和IKKβ参与信号传递,DDX3不但参与信号传递,还是SGs组装过程中必不可少的蛋白。本研究进一步对在SeV感染过程中,这些信号蛋白是否迁移到SG进行检测。以1 MOI SeV感染HeLa细胞,设置模拟感染为对照组。感染后16 h,利用G3BP1鼠抗和TAK1兔抗、G3BP1鼠抗和IKKβ兔抗,或G3BP1鼠抗和DDX3兔抗进行免疫荧光实验,检测G3BP1和NF-κB信号通路的蛋白激酶TAK1、IKKβ、或RNA解旋酶DDX3的亚细胞定位。如图3所示,在模拟感染的细胞中,G3BP1均匀分布于细胞质,没有观察到SG的形成;蛋白激酶TAK1、IKKβ,或RNA解旋酶DDX3也均匀分布于细胞质和细胞核;在SeV感染的一部分细胞中,G3BP1形成点状聚集,形成SGs;TAK1、IKKβ或DDX3也形成点状聚集,跟G3BP1共定位。以上结果说明SeV感染诱导TAK1、IKKβ和DDX3聚集于SGs。

图3 TAK1、DDX3和IKKβ的免疫荧光共聚焦实验Fig.3 Confocal immunofluorescence assay of TAK1、DDX3和IKKβ
2.4 泛素化酶和去泛素化酶A20以及去泛素化酶CYLD在SeV的感染过程中聚集于SGA20,也被称为肿瘤坏死因子诱导蛋白3(tumor necrosis factor induced protein 3, TNFIP3),具有泛素连接酶和去泛素化酶的功能,由NF-κB诱导表达,对RIP1和其他信号转导蛋白进行泛素化或去泛素化,负向调控NF-κB信号的强度和持续时间,形成负反馈回路[38-39]。A20阻止了IFN和细胞因子的大量表达,对免疫系统的稳态至关重要。CYLD(cylindromatosis)是一种去泛素化酶,属于去泛素化酶USP家族(ubiquitinspecific proteases),通过其C末端的泛素水解结构域,可以特异性的从蛋白底物上除去K63连接的泛素链,从而改变靶蛋白的功能[40-42]。同时,CYLD还具有一个特异性位点,可以与肿瘤坏死因子受体相关因子2(tumor receptor-associated factor 2, TRAF2)和NF-κB信号通路激酶((NF)-kappaB essential modulator, NEMO)结合[43]。因此,A20和CYLD在NF-κB号通路的调节中发挥着重要的作用[44]。我们接下来对A20和CYLD进行检测,观察其在SeV感染过程中亚细胞定位的变化。以1 MOI SeV感染HeLa细胞,设置模拟感染为对照组。感染后16 h,利用G3BP1鼠抗和A20兔抗,或用G3BP1鼠抗CYLD兔抗进行免疫荧光实验,检测G3BP1和泛素化相关酶A20或CYLD的亚细胞定位。如图4所示,在模拟感染的细胞中,G3BP1均匀分布于细胞质,没有观察到SG的形成;A20和CYLD也均匀分布于细胞质和细胞核;在SeV感染组,一部分细胞中的G3BP1形成点状聚集,形成SGs;A20和CYLD形成点状聚集,跟G3BP1共定位。以上结果说明SeV感染诱导SGs的形成,参与信号传导的A20和CYLD聚集于SGs。

图4 A20和CYLD的免疫荧光共聚焦实验Fig.4 Confocal immunofluorescence assay of A20和CYLD
2.5 A20及其磷酸化形式p-A20在poly I:C刺激下聚集于SG如前所述,在SeV感染Hela细胞的过程中,很多与天然免疫相关的信号蛋白会聚集到SG。然而,信号蛋白聚集到SG是否为SEV感染细胞特有的现象,尚不清楚。poly I:C是病毒双链RNA模拟物,能够通过激活PKR,刺激SG的形成,并激活干扰素信号通路。因此,poly I:C刺激能够模拟RNA病毒感染。我们将探讨在poly I:C刺激细胞后,天然免疫信号蛋白是否聚集于SG。我们接下来以A20及磷酸化活化的A20(p-A20)为代表性蛋白进行检测和分析。在HeLa细胞中转染poly I:C,设置PBS转染为对照组。转染后16 h,利用G3BP1鼠抗和A20兔抗或p-A20兔抗进行免疫荧光实验,检测G3BP1和A20及p-A20的亚细胞定位。如图5所示,在PBS刺激的细胞中,G3BP1均匀分布于细胞质,没有观察到SG的形成;A20均匀分布于细胞质和细胞核;在poly I:C刺激的细胞中,G3BP1形成点状聚集,形成SG;一定比例的A20和p-A20形成点状聚集,跟G3BP1共定位。以上结果说明poly I:C刺激能诱导SG的形成,参与信号传导的A20及其活性形式p-A20聚集于SG。以上结果揭示,信号蛋白聚集到SG并不是SEV感染特有的现象。
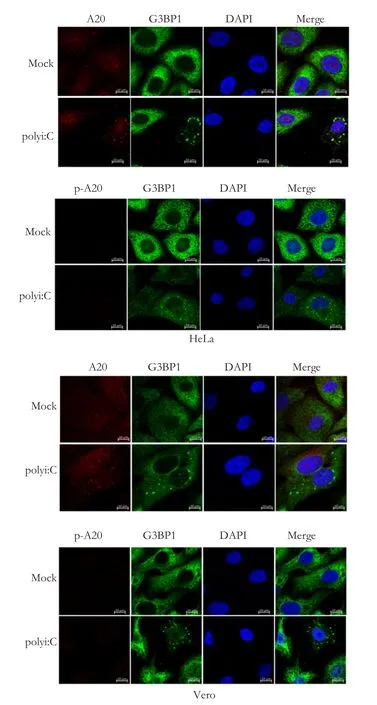
图5 A20和p-A20的免疫荧光共聚焦实验Fig.5 Confocal immunofluorescence assay of A20和p-A20
3 讨论
在病毒感染细胞时,天然免疫应答是宿主的第一道抵御防线。干扰素是天然免疫应答的关键抗病毒蛋白,由PRRs识别病毒感染后,激活IRF3/IRF7或NF-κB诱导表达。病毒感染产生的双链RNA,结合并激活PKR,PKR进一步磷酸化eIF2α,使蛋白翻译所需的三元复合物eIF2-GTP-Met-tRNAMet不能循环利用,细胞迅速终止蛋白翻译,在细胞质中形成的致密颗粒状聚合体SG,临时储存mRNA和翻译元件。已有的研究显示,RIG-I和PKR在NS1缺失的禽流感A型病毒(Influenza A virus, IAV)的感染过程中,迁移到SG,SG发挥促进干扰素反应的作用[45],因此,SG在IAV的感染中,具有抗病毒作用。IAV进化出干扰SG形成的机制:非结构蛋1(NS1)通过抑制PKR的激活,避免磷酸化eIF2α,从而不诱导SG的形成;而NS1缺失型IAV(IAV-NS1),则显著地诱导依赖于PKR-eIF2α途径的SG形成[3,45]。有报道显示:脊髓灰质炎病毒(Poliovirus, PV)和脑心肌炎病毒(Encephalomyocarditis virus, EMCV),这两种病毒能暂时诱导SG形成,但是病毒3C蛋白具有切割SG核心蛋白G3BP1的功能,从而抑制SG形成[19,46]。有研究表明,SeV通过C蛋白减少双链RNA的累积,逃逸对PKR的激活和对SG的诱导。C蛋白缺陷型SeV有效刺激SG的形成,没有核衣壳的病毒RNA迁移到SG。SeV具有拮抗SG的机制,那么,SG在SeV感染过程中,是否发挥抗病毒作用呢?
在本研究中,我们发现SeV感染HeLa细胞的过程中,大部分感染的细胞不形成SG,一小部分细胞形成SGs。RNA感受器MDA5和RIG-I,下游的蛋白激酶如TBK1、IKKε、TAK1,IKKβ、RNA解旋酶DDX3、泛素化酶和去泛素化酶A20以及去泛素化酶CYLD均聚集到SG。结合前人的研究,我们推测在SeV感染细胞的过程中,病毒RNA被招募到SGs,PRRs迁移到SGs,识别病毒RNA,把信号传递给下游的激酶,最终激活IRF3/7和NF-κB,诱导干扰素反应。因此,我们预测在SeV感染过程中,SG是宿主识别病毒感染并发生信号传递的一个平台,SeV通过C蛋白拮抗SG的形成,减少宿主细胞的抗病毒反应。
天然免疫相关的信号蛋白聚集到SG,并不是SeV感染特有的现象。在我们的研究过程中,发现poly I:C刺激诱导的SG,也具有PRRs和A20等天然免疫相关蛋白。并且,G3BP1/2缺失的细胞系,在poly I:C刺激下,不能形成SG,干扰素反应明显下降。因此,SG在病毒感染过程中作为一个启动或增强天然免疫应答的信号平台,发挥抗病毒作用[47]。本研究证实了在病毒感染细胞的过程中,信号蛋白聚集到SG是一种普遍现象,为进一步明确SG在抗病毒反应中发挥的作用提供参考,同时揭示细胞压力应激与干扰素反应存在交叉联系。
猜你喜欢
课外生活·趣知识(2022年2期)2022-02-08
医学综述(2020年11期)2020-02-16
中学生数理化·高二版(2017年3期)2017-07-07
电脑知识与技术(2016年31期)2017-02-27
科学中国人(2016年30期)2016-07-14
建材发展导向(2016年3期)2016-05-23
中国蔬菜(2015年9期)2015-12-21
浙江大学学报(农业与生命科学版)(2015年4期)2015-12-15
应用海洋学学报(2015年3期)2015-11-22
中国医学科学院学报(2015年5期)2015-03-01
