
SuperLLEC:全新的链读和长读测序组装纠错算法
2022-02-24 12:35崔雅轩张少强
计算机工程与应用 2022年3期
崔雅轩,张少强
天津师范大学 计算机与信息工程学院,天津 300387
1977年诞生的第一代DNA(deoxyribo nucleic acid,脱氧核糖核酸)测序技术(Sanger测序)[1]直接推动了2000年人类基因组计划的完成[2]。Sanger测序的读长达到1 000 bp(base pairs,碱基对),精准度达到99%,但该测序技术通量低且成本高昂[2]。为了降低成本和提高通量,第二代测序技术应运而生,第二代测序技术主要包括ABI公司的SOLID测序技术,Roche/454的GS FLX和Illumina公司的Solexa Genome Analyzer[3]。第二代测序一般产生长为75~500 bp的短读(short-reads)。为了在高通量下提高测序读长,近几年第三代长读(long-reads)测序技术开始大量应用,最具代表性是太平洋生物科学公司(PacBio)的单分子实时测序(single molecule real time,SMRT)[4]和牛津纳米孔技术公司(Oxford Nanopore)的纳米孔测序[5]。相较于前两代测序技术,第三代测序具有无需PCR(聚合酶链式反应,polymerase chain reaction)扩增和读长更长(一般10 000~30 000 bp)的特点,但测序准确度有所降低(准确率在85%左右)[6-8]。
由于第三代测序较高的错误率,为了提高序列组装的准确度,长读测序的基因组组装算法往往需要与纠错算法进行合作。目前基于第三代测序的基因组组装算法主要有Wtdbg2[9]、FALCON[10]和Canu[11]等;这三个算法均能够基于单体型进行组装,识别其中的单核酸多态性(single nucleotide polymorphism,SNP)位点。而目前第三测序的纠错算法主要分为两类[12]:一类是第三代数据自纠,这类最新算法主要有FLAS[13]、LoRDEC[14]等;另一类是借助第二代数据对第三代数据进行纠错,例如基于序列比对的纠错算法proovread[15]和CoLoRMap[16];以及基于构建de Bruijn图的纠错算法FMLRC[17]和Par-LECH[18]等。而PacBioToCA算法既能对PacBio测序数据自纠,也能用二代Illumina测序数据对PacBio数据进行纠错[19]。
上述纠错算法均采用先纠错再序列组装的策略,但2017年10xGenomics公司对Illumina二代测序方案进行升级,引入条码(barcode)标签序列,相同条码的短读集合形成跨度在30 000~100 000 bp的链读(linked-reads)信息[20]。为了充分运用序列跨度大的链读数据关联信息,和基于最新的第三代测序组装算法性能的大幅提升,本研究放弃先对长读纠错再组装的策略,拟采用先组装再纠错和进一步组装的策略:先对PacBio长读组装得到的支架序列(scaffolds),再利用带条码标签的链读测序数据设计了对支架进行纠错和进一步组装的算法SuperLLEC,由此得到更高质量的基因组单体型序列。
1 数据描述
1.1 10X Genomics链读
10X Genomics公司通过在序列中引入条码(barcode)标签序列[20],即一段固定长度的碱基短序列,并将其分配到不同的油滴微粒(droplet)中,利用GemCode平台对带条码标签的长片段序列进行扩增,最后将序列打碎成合适测序短片段进行二代测序。通过条码标签序列信息追踪来自每个长片段DNA模板的多个读序(read),称为链读(linked-reads),从而获得该长片段的大量短读序列。为了方便描述,本实验将具有相同条码标签的所有链读序列合称为“链读序列组”。同一链读序列组的所有读序均来自相同的染色体(跨度为30 000~100 000 bp)。此信息有助于对组装后的支架序列根据链读相关性进行进一步拼接。研究所用链读数据均从10X Genomics官网下载(https://support.10xgenomics.com/de-novo-assembly/datasets)。
1.2 PacBio长读
PacBio长读序列,是太平洋生物公司的单分子实时测序技术平台产生的测序读序[21]。长读序列的单一测序长度一般大于10 000 bp[22]。实验用到的PacBio数据集来自三种人类基因组样本的测序数据,具体信息见表1,数据下载地址见表2。

表1 实验所需PacBio长读数据Table 1 PacBio long read data in experiment

表2 实验所需PacBio长读数据下载地址Table 2 Download site of PacBio long read data
2 方法和步骤
算法主要分三步:首先对PacBio三代测序数据用组装算法组装为支架序列;然后将相同条码标签的链读序列组快速分配给支架集合,再用序列比对工具将所有链读一一比对到对应的支架上,并根据链读信息对支架继续组装;最后,统计多态位点的碱基频率来判断是否为测序错误碱基或者是SNP。如果是测序错误碱基,则根据位点频率进行纠错以提高基因组拼接质量。其中在链读与支架比对过程中,可以对链读集合进行手动拆分多个子集后多核并行化快速处理(文中用8个进程进行实验)。图1是算法的研究方法流程示意图。下面就算法各步骤进行详细阐述。

图1 研究方法流程示意图Fig.1 Schematic flowchart of research method
2.1 组装长读数据构建支架序列
对原始的PacBio测序数据,首先用第三代测序的基因组组装算法将其拼接成支架序列集合。Wtdbg2[9]、FALCON[10]、Canu[11]是三种最新的第三代测序组装算法。实验分别尝试使用这三种算法依次对表1的三组长读数据集进行序列组装。对组装完成的数据进行比较分析后发现,Canu组装后的人类基因组数据规模(约为3.42 GB)大于Wtdbg2和FALCON的数据规模(约为3 GB)。另外实验发现Wtdbg2在三者中是运算最快的算法。例如对于Human CHM1的PacBio长读数据,Canu的CPU用时为22 750 h,FALCON用时为68 789 h,而Wtdbg2用时只有245 h。这是因为Wtdbg2算法采用Linux的Pthreads多线程处理技术来加快算法的执行速度。由此,实验采用Wtdbg2算法对实验的PacBio长读数据组装成支架序列集合。在运行Wtdbg2时,除了记录支架序列外,还记录下碱基多态位点(Wtdbg2预测为SNP)的碱基频次(比对到该位点的长读碱基频次)。
2.2 链读序列组分配给支架
对每个支架序列S,以k为步长,将其拆分成长度为k的短序(称为k-mer),若支架序列S长度为L,则可以得到L/k个k-mer;并将得到的所有k-mer存储在哈希表中,哈希表的键为k-mer,键值为该k-mer所在的支架编号S。对每组链读序列中的读序,以长度k为窗口,以步长1遍历所有的k-mer,并通过哈希表确定该组链读序列的k-mer存在于支架的编号集合,据此将每组链读序列集合分配给支架。如图2示例所示,通过判断链读和支架是否共享k-mer,将四组链读序列集合依次分配给两条支架序列。其中Barcode编号为2的链读组同时分配给了两条支架。

图2 纠错组装示意图Fig.2 Flowchart of assembly and error correction
为了加快运算速度,实验将所有链读序列组平均分成8组,调用8个线程分别进行支架分配。
2.3 链读序列比对到支架并进一步组装
将分配到每个支架的每组链读短序比对到该支架序列上。由于经典对比工具BLAST[23]采用的是近似比对算法,适用于长序列之间的快速比对,不适用于短序列比对。实验采用Bowtie2[24]进行序列比对。Bowtie2具有比对速度快和占用内存低的优点。
对每组具有相同条码标签的链读序列,用Bowtie2工具把它们比对到与之关联的支架上。如果某组链读序列之前被分配给多个支架,则根据该组链读条码信息和重叠关系,将这些支架进一步拼接成超级支架(superscaffold)序列。如图2示例所示,A和B两个支架序列因为Barcode 2而被拼接成一条超级支架序列。
为了加快运算速度,实验将所有支架序列分成8组,保证每组之间没有共享的链读序列集,分别进行序列比对。同时,对于比对出现多态(或称不保守)的位点,分别记录链接读序在该位点出现的碱基频次。例如,如图3示例所示,在标出的第一列位置,碱基C在对比上的链读中出现3次。

图3 比对后的碱基频率示意图Fig.3 Example of base frequency statistics after alignment
2.4 纠错和SNP预测
算法最后采用Fisher精确检验(Fisher’s exact test)来检验每个位点碱基是否有错误碱基或者SNP。Fisher精确检验是一种分析列联表(contingency table)统计显著性检验方法,用于检验两个分类的关联度。
如果某个位点只有一种碱基,则无需计算,说明该位点碱基信息一致;否则,在支架对应位点上,统计比对后的链读出现频次最高的两个碱基X和Y(如图3示例,支架在碱基不一致的T/G位点,该位点链读出现频次最高的碱基是T和C),并记录频次为clong和dlong,在支架上这两个碱基出现的频次为alinked和blinked。注意如果在支架序列出现不是X或Y的第三种碱基Z,根据链读的精确性,自动认定碱基Z为测序错误的位点,不考虑Z出现的频次。为了保证假设检验碱基样本数量的一致性,分别计算相对频次c=[100clong/(clong+dlong)],d=[100dlong/(clong+dlong)],a=[100alinked/(alinked+blinked)],b=[100blinked/(alinked+blinked)]。如表3所示,为了比较长读组装和链读比对的差异,构造长读和链读碱基频次的列联表,然后采用Fisher精确假设检验。对一个位点,Fisher精确检验的零假设为长读和链读在该位点的相对频次没有差别。运用超几何分布计算Fisher精确检验的P值(P-value):


表3 某位点在长读和链读碱基频次列联表Table 3 Contingency table of site located by long reads and linked-reads
为了快速计算P值,公式(1)中的阶乘均可以采用公式(2)的斯特林(Stirling)公式来近似计算,由此得到取对数的组合数的近似计算公式(3),并通过公式(4)防止数据溢出。

对于某位点,如果计算的P值大于等于设定的临界值0.05,则判定零假设成立,该位点在链读中出现频率最高的两个碱基预测为SNP;如果P值小于临界值,则判定零假设不成立,那该位点在链读中出现频率较小的碱基则判定为测序错误的碱基。
3 实验与结果分析
3.1 实验环境
实验所有数据均在CPU为Intel Xeon E2244G 3.80 GHz 8 Cores,内存为512 GB的CentOS系统服务器上调试运行。
3.2 结果分析
针对三组人类基因组样本PacBio测序数据集(Human NA24385、Human HG00733、Human CHM1分别记为Data1、Data2、Data3),分别用PacBioToCA和LoRDEC对这三组PacBio测序数据集进行自纠;另外也基于10X Genomics的链读数据分别运行PacBioToCA和proovread对这三组PacBio数据集进行纠错;最后对纠错后的三组数据用Wtdbg2算法进行基因组组装。
针对SuperLLEC算法,实验是先用Wtdbg2算法分别对这三组PacBio测序数据组装成支架序列(其运行时间和输出支架总规模见表4),然后运用10X Genomics的链读数据用SuperLLEC对支架进行纠错和进一步组装。

表4 Wtdbg2运行时间和输出规模Table 4 Running times and result sizes of Wtdbg2 on three datasets
支架NG50[7]是在基因组组装中衡量算法优劣的重要指标之一,其代表含义是算法所组装得到的支架序列的中位长度。如表4所示,在没有运用任何纠错算法的情况下,Wtdbg2对三个数据集进行组装,其支架NG50长度分别为25.8 MB、12.7 MB和15.4 MB。而如表5所示,在运用PacBioToCA、LoRDEC和proovread进行纠错后,再用Wtdbg2组装,如表5所示,这三个数据集的支架NG50增长不明显,三个工具中最大的分别为26.1 MB、13.1 MB和16.2 MB。除了PacBio数据自纠外,这三种算法均用到了链读数据,但是这三种算法只把链读单纯作为独立的短读,没有用到链读条码关联信息。但是从表5可以看出SupperLLEC组装得到的支架NG50长度分别为28.3 MB、16.1 MB和17.5 MB,远远大于其他几个先纠错再组装的算法实验。进一步合并三个数据集的结果,图4分别画出Wtdbg2、SuperLLEC和其他所有工具的支架长度分布,发现SuperLLEC与Wtdbg2和其他工具之间有显著差异,其大尺寸的支架占比更大。因为SuperLLEC充分利用链读数据的关联关系进行了进一步的支架组装,而其他纠错算法无法运用链读数据的条码信息,所以其他纠错算法无法达到SuperLLEC算法的组装长度。

图4 数据集纠错前后支架序列长度分布比较Fig.4 Length distributions of scaffolds before and after error corrections

表5 NG50长度对比表Table 5 Comparison of NG50 length
另外,通过和NCBI RefSeq真实基因数据集比较,如表6所示,在组装准确率上,SuperLLEC在三个数据集上均明显优于其他纠错算法,达到了非常高的准确率。同时,因为其他纠错算法是先纠错后读序组装,没有单独SNP的预测,其SNP预测均来自Wtdbg2的预测,因此在SNP覆盖率(即召回率,预测正确的SNP数除以SNP总预测数)上其他纠错算法没有明显变化。如表7所示,在三个数据集上,SuperLLEC的SNP覆盖率均超过80%,大大提升了Wtdbg2的召回率。因此SuprLLEC可以与Wtdbg2结合使用来提高组装的准确率和SNP预测精度。
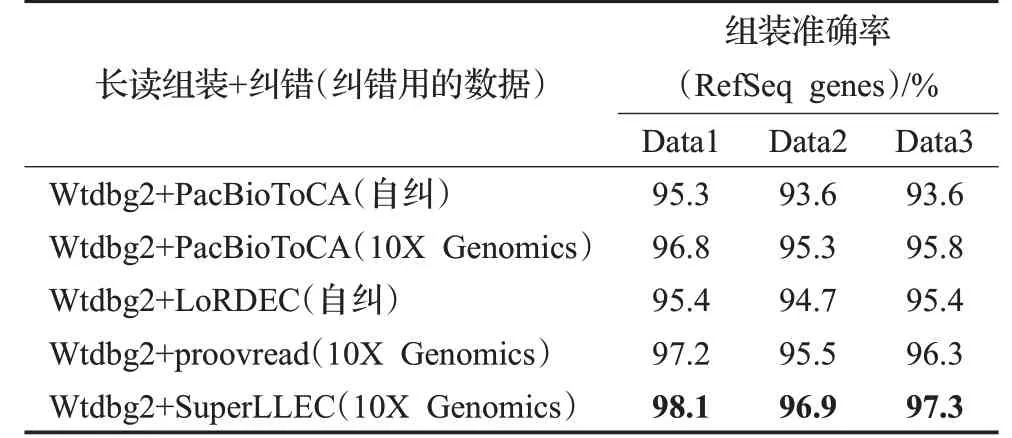
表6 组装准确率对比表Table 6 Comparison of assembly accuracy
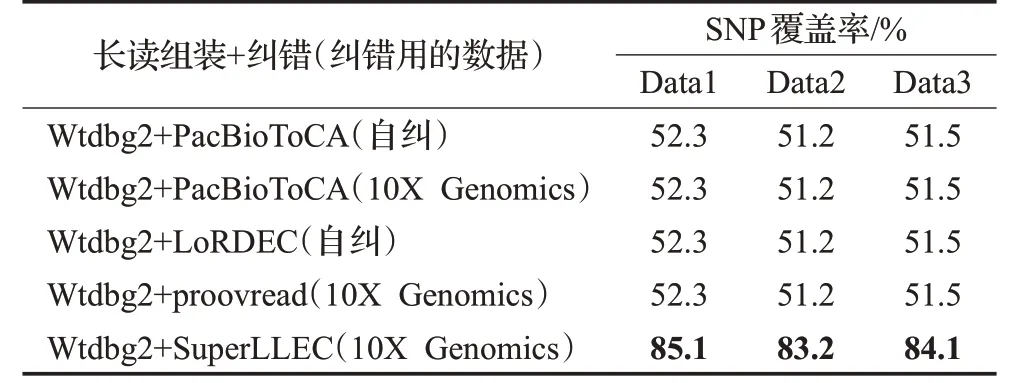
表7 SNP覆盖率对比表Table 7 Comparison of SNP coverage
4 结束语
SuperLLEC算法是借用跨度大的链读数据集对PacBio三代测序数据进行纠错和组装。SuperLLEC算法有别于传统的纠错算法:首先,放弃直接纠错,而采用先组装后纠错,并且在纠错过程中进一步组装。其次,根据链读及其条码信息与PacBio数据结合进行组装和纠错,目前主要算法均为第三代数据自纠或运用第二代测序短读数据进行纠错,因此只能借用短读的双末端(pair-end)信息,而SuperLLEC首次根据链读条码信息进行纠错和组装,组装和纠错效果更好。再次,算法采用的Fisher精确检验比经典的卡方检验更适合离散分布的检验,能够更好地辨别单核酸多态性碱基和测序错误碱基。最后,算法步骤中多次运用并行计算技巧加快算法的运算速度。目前由于第三代测序技术还存在测序准确率偏低的问题,因此该纠错组装算法能够很好地提高基因组组装的准确率。
猜你喜欢
条码与信息系统(2021年1期)2021-12-05
今日农业(2021年11期)2021-08-13
中国生殖健康(2020年4期)2020-12-09
商品与质量(2020年46期)2020-11-26
教学考试(高考生物)(2020年6期)2020-11-23
中西医结合肝病杂志(2020年2期)2020-10-27
条码与信息系统(2020年5期)2020-06-07
食品与生物技术学报(2020年8期)2020-01-06
学苑创造·B版(2019年5期)2019-06-14
科学24小时(2019年5期)2019-06-11
