
自然群体多性状表型缺失值预测方法的比较
2022-04-01 12:10王媛温阳俊王艳萍刘汉钦马若洵吴清太张瑾
南京农业大学学报 2022年2期
王媛,温阳俊,王艳萍,刘汉钦,马若洵,吴清太,张瑾
(南京农业大学理学院,江苏 南京 210095)
缺失数据是影响数据分析准确性的重要原因之一,完整表型和基因型数据的分析对防治疾病、遗传育种等研究具有重要意义。目前,在全基因组关联研究(genome-wide association study,GWAS)中基因型数据的缺失已经有了相对成熟的处理方法[1-5],而表型数据的缺失直接减少样本量,削弱GWAS的统计推断能力。表型的缺失值主要有3种处理方法:直接删除法、重测量法和插补法。直接删除法将数据中含有缺失值的样本点全部删除,从而得到一个不含缺失值的“完整”数据集。由于被删除的样本中可能包含重要信息,直接删除会造成原始信息的损失,减少样本量,降低后续统计推断的功效。重测量法,即进行重复试验,采集相同条件下的数据,对原有缺失值进行插补。虽然重测量法能够插补缺失的数据,但耗费大量的人力和物力。数据插补法利用现有观测数据信息,借助统计学方法预测缺失值以产生完整数据[6],插补表型缺失避免了原始信息的损失、增大了样本量,具有省时高效的优点。
精确的缺失表型预测可以提高后续统计分析的准确性,常用的方法有:均值插补法(mean)[7],利用观测数据的均值插补缺失值,简单易行,但是扭曲了样本的分布,降低了数据的变异程度;回归插补法[8],利用完整的观测数据建立回归模型,依据此模型预测表型缺失值;多重插补法[9],对缺失数据进行多次插补得到多组完整数据集,通过统计方法和综合分析推断出一组最佳完整数据集;EM算法[10],根据观测数据的边缘分布,利用EM算法进行极大似然估计,对含有缺失数据的不完整数据进行预测,但数据量大时收敛速度慢、计算繁琐,效果不佳。
近年来,机器学习算法在缺失数据预测插补中也得到了应用[11]。K近邻(K-nearest neighbor,KNN)插补法[12],将最邻近的k个样本观测值的加权平均值作为缺失数据的估计值,其插补效果易受异常值的影响,产生误差较大;支持向量机[13]将完整数据集作为训练集,获得预测模型后插补缺失数据,大样本情况下的插补精确度高;随机森林[14]利用bootstrap抽取k个样本集,组成k个决策树,利用全部决策树构建的随机森林插补缺失值;神经网络[15]将误差反向传播来训练多层网络,通过优化网络输出不断减小误差,在随机缺失数据插补中应用广泛。机器学习算法在处理数据缺失问题时速度快、泛化能力强,但由于训练数据对参数影响较大,插补能力还有待提高。此外,基于混合线性模型的缺失值插补方法[16-19]进一步提高了表型缺失值插补能力。
与单性状插补方法相比,多性状联合插补分析可以利用性状之间的遗传结构[20],并对多个性状缺失值同时进行插补,省时高效。多重插补法(multiple imputation by chained equations,MICE)[9],利用观测数据进行多变量回归,以回归的估计值插补缺失值,由于每个插补值都要进行多重插补计算,插补时间较长;PHENIX(phenotype imputation expediated)插补法[21]建立在贝叶斯多表型混合模型上,在亲缘关系已知的情况下利用变分贝叶斯方法进行拟合;softImpute插补法[22]是一种利用核范数(或奇异值)正则化来插补矩阵缺失值的交替迭代算法,利用核范数定义损失函数并利用奇异值分解求解核范数使损失函数最小,精度高、速度快;多表型混合模型(multi-phenotype mixed models,MPMM)[16]是一种基于多个相关性状的混合模型方法,利用其条件期望或最优线性无偏预测对缺失的数据进行插补,具有精确度高的特点。多性状基因组模型(multiple-trait genomic model,MTGM)[23]在单性状模型的基础上进行拓展,可应用于估计育种值的预测问题。多性状联合插补方法是在利用变量之间关系的前提条件下对缺失值进行预测,结果往往比较准确,通常会明显优于单变量插补方法[24]。多表型插补技术已经成功应用于人类[20]、酵母、鼠、鸡、小麦和大豆[25]等多个物种的缺失数据集上,有效提高了缺失数据的预测精度,进而提升全基因组关联分析的计算效率和准确性。
表型缺失数据插补的准确性受到缺失率、样本量等因素的影响,上述方法尚未针对这些因素对插补效果进行评判。本研究利用mean、KNN、决策树(decision tree,DT)[26]、MICE、PHENIX和softImpute插补方法对多表型模拟缺失数据进行预测,以均方误差(mean squared error,MSE)、皮尔逊相关系数(Pearson correlation coefficient,r)和计算时间为指标,比较在不同表型缺失率(phenotypic missing rate)、性状数(D)、样本量(n)和性状相关性(cor)下的插补效果。使用6种方法对拟南芥实际数据的长日照花期(days to flowering under long day)、短日照花期(days to flowering under short day)、春化长日照花期(days to flowering under long day with vernalization)和春化短日照花期(days to flowering under short day with vernalization)的表型缺失值进行多性状联合插补,并对插补后的完整数据进行全基因组关联分析,通过基因功能验证插补数据的可靠性,查找出与目标性状关联的已验证基因。
1 材料与方法
1.1 模拟试验材料
基于线性模型,生成等位基因频率为[0.1,0.5]的10 000个单核苷酸多态性(single nucleotide polymorphism,SNP),随机设置10个数量性状核苷酸(quantitative trait nucleotide,QTN),其总遗传率是60%,正态误差方差设置为1.0。在此基础上,设计了模拟试验(表1)。
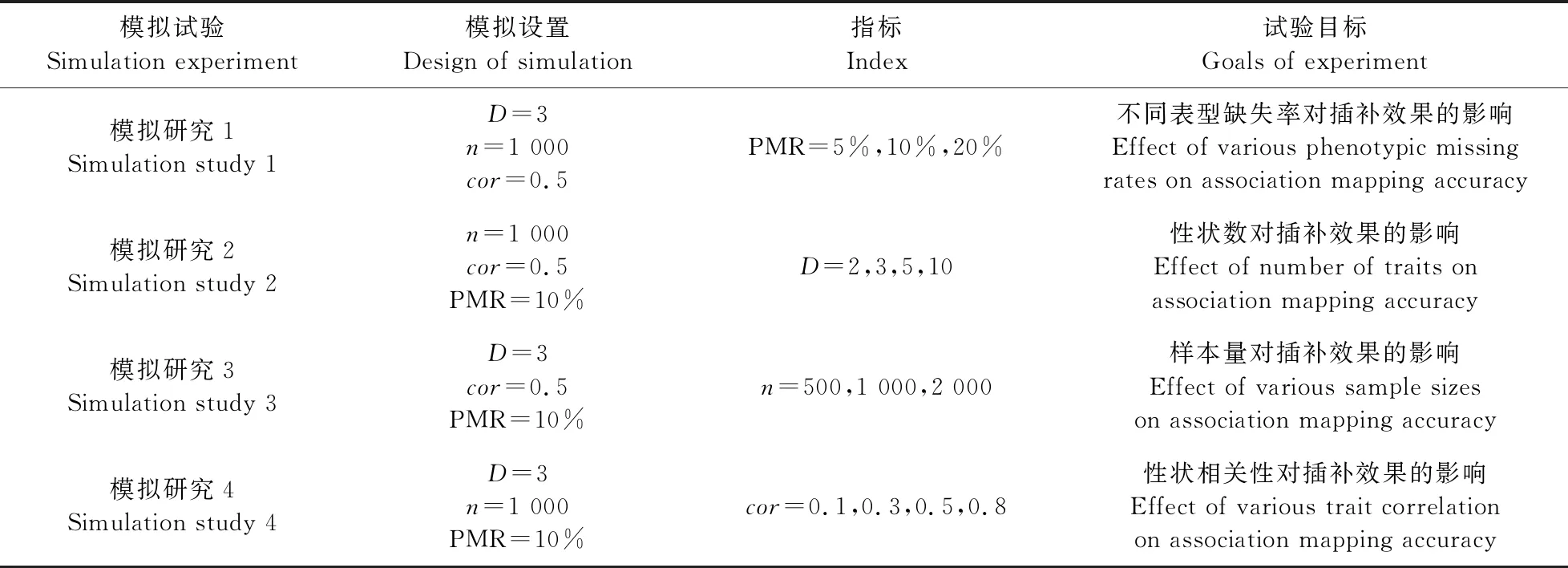
表1 4个模拟研究设计
利用表型数据插补方法对上述缺失数据进行预测,使用MSE、r和计算时间等指标对插补数据的准确性进行评判。
1.2 真实试验材料
真实数据集[27]来自拟南芥自然群体的199个个体,216 130个SNP(http://www.arabidopsis.usc.edu/),考虑长日照花期、短日照花期、春化长日照花期和春化短日照花期4个花期相关性状(https://www.arabidopsis.org/portals/genAnnotation/index.jsp),其表型缺失率分别为16.0%、18.6%、15.6%和 20.1%。利用mean、KNN、DT、MICE、PHENIX和softImpute插补方法对缺失数据进行预测,并对插补后的完整数据集进行全基因组关联分析得到显著SNP,利用TAIR基因库(https://www.arabidopsis.org)验证位于显著SNP附近20 kb的关联基因,挖掘出与花期相关的已验证基因。
1.3 插补方法
1)mean插补法[7]:用当前变量的所有观测值平均值代替其所有缺失值的方法。2)KNN插补法[12]:首先选出与缺失值距离最近的k个样本观测数据,根据其距离加权计算出缺失数据的估计值,用该值来插补对应的缺失值。KNN插补法基于R软件程序包DMwR2实现,k默认设为10。3)DT[26]插补法:利用观测数据集构建基于回归树的DT模型,然后将含有缺失值的数据代入DT模型进行插补。DT插补法基于R软件程序包rpart实现。4)MICE插补法[9]:是一种重复计算缺失值的处理方法。它首先多次计算缺失值,并产生m个完整数据集;然后对这m个插补数据集进行统计分析,得到m个分析结果;最后综合m个分析结果做出统计推断,最终得到缺失值的最优估计值。综合起来,其主要步骤可总结为:插补、分析、综合。MICE插补法基于R软件程序包mice实现。5)PHENIX插补法[21]:充分利用表型之间的相关性和亲缘关系矩阵,基于贝叶斯多表型混合线性模型,利用变分贝叶斯方法对模型进行拟合,根据观测数据的后验多变量正态分布得到缺失数据的近似估计值。PHENIX插补法基于R软件程序包phenix实现。6)softImpute 插补法[22]:将矩阵完备化问题转化为核范数最小二乘问题,利用软阈值奇异值估算矩阵的缺失值。该算法的计算原理类似于EM算法,每一次迭代,首先通过对估计矩阵的核范数(或奇异值)的惩罚来估计低秩表型矩阵的近似矩阵,并利用估计值对缺失矩阵进行插补,然后利用软阈值奇异值分解法,向目标函数的最小值逼近,得到近似最优解的集合,不断迭代以对完整矩阵进行不断优化。softImpute插补法基于R软件程序包softImpute实现。
1.4 评估指标
采用均方误差MSE和皮尔逊相关系数r来衡量基于不同表型缺失率、性状数、样本量和性状相关性的插补准确性。MSE计算公式为:
(1)
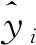
(2)

2 结果与分析
2.1 模拟数据的缺失数据插补
利用mean、KNN、DT、MICE、PHENIX和softImpute插补方法对模拟缺失数据进行预测。针对不同表型缺失率、性状数、样本量和性状相关性,比较各个方法插补效果的影响。
模拟研究1(表1)考察缺失率分别为5%、10%和20%时缺失数据的插补效果。结果表明,随着表型缺失率的增大,6种方法的MSE(图1-A)不断增大,r不断减小(图1-B),这意味着缺失值插补的准确性随着表型缺失率的增大而降低,这与事实相符。其中,PHENIX插补法准确性最优,随着缺失率的增大,该方法在插补准确性上的优势变得越来越明显,说明PHENIX插补法更适用于高缺失率的情形。KNN和DT插补法的准确性比PHENIX略微逊色,其次是softImpute和mean插补法,而MICE准确性受表型缺失率的影响最明显,尤其在高缺失率的情况下表现不佳。
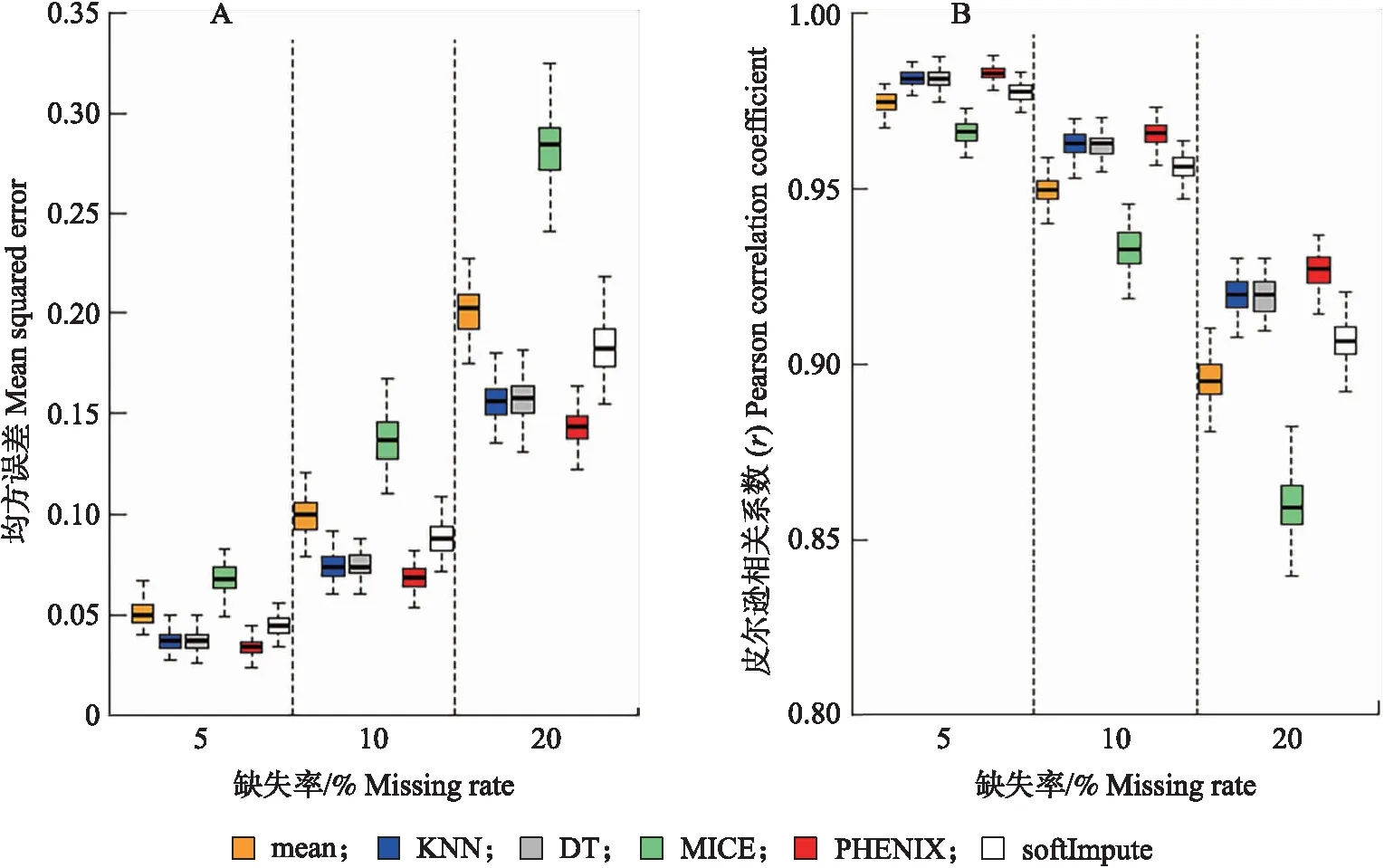
图1 不同表型缺失率下mean、KNN、决策树(DT)、MICE、PHENIX和softImpute插补值与观测值的均方误差(A)和皮尔逊相关系数(B)(重复100次)
模拟研究2(表1)考察性状数(D)为2、3、5和10时缺失数据的插补效果。mean插补法的效果不受性状数的影响,其他5种方法随性状数的增大,MSE(图2-A)不断减小,r不断增大(图2-B),说明上述多表型缺失数据插补方法均能够利用多性状之间的相关性和潜在信息,性状数不断增加,插补的准确性不断提高。其中,PHENIX的插补效果最优,其次是DT和KNN,softImpute的插补效果随性状数的增加逐渐优化,这4种插补方法的效果均优于mean和MICE插补法。

图2 不同性状数下mean、KNN、DT、MICE、PHENIX和softImpute插补值与观测值的均方误差(A)和皮尔逊相关系数(B)(重复100次)
模拟研究3(表1)考察样本量(n)为500、1 000和2 000时缺失数据的插补效果。在相同表型缺失率(10%)下,样本量的增大对于插补精确度没有显著影响,但是MSE和r的箱线图方差变小(图3),说明样本量越大,结果越稳定。趋势与上述模拟试验相似,依然是PHENIX插补法的效果最好,其次是KNN和DT插补法,这3种插补方法的效果均优于softImpute、mean和MICE插补法。
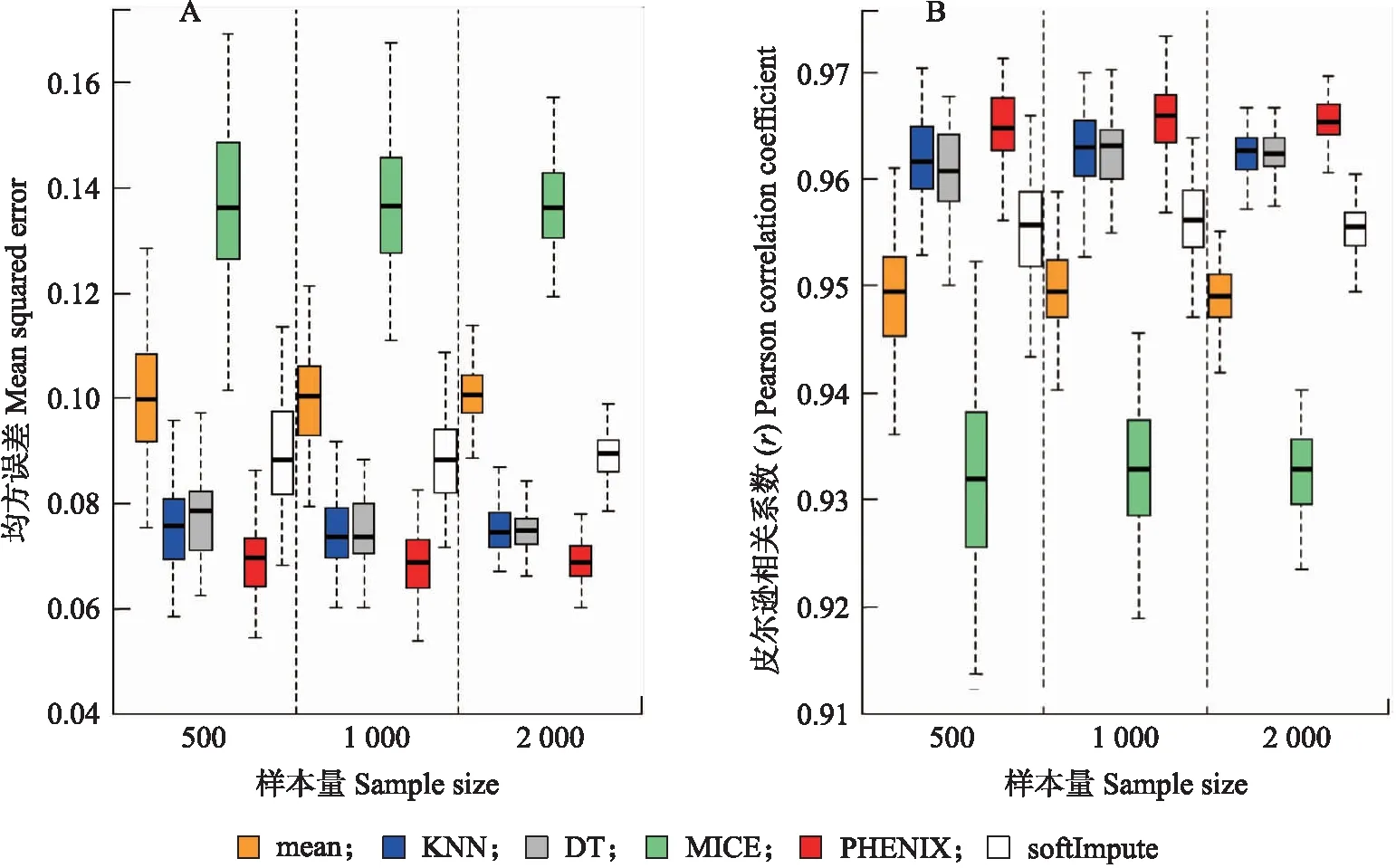
图3 不同样本量下mean、KNN、DT、MICE、PHENIX和softImpute插补值与观测值的均方误差(A)和皮尔逊相关系数(B)(重复100次)
模拟试验4(表1)考察性状相关性(cor)为0.1、0.3、0.5和0.8时缺失数据的插补效果。结果表明mean插补法的效果不受性状之间相关性强弱的影响,而其他5种方法随着性状之间相关性的增加,MSE(图4-A)不断减小,r不断增大(图4-B)。这意味着性状之间的相关性越强,KNN、DT、MICE、PHENIX和softImpute方法越能够利用相似的遗传结构,插补效果越好。在弱相关的情况下,softImpute方法和KNN、DT、PHENIX插补法的效果相似,具有较高的准确性,随着性状相关性的增大,softImpute插补准确性与它们差距变大,上述方法的准确性均优于mean和MICE插补法。
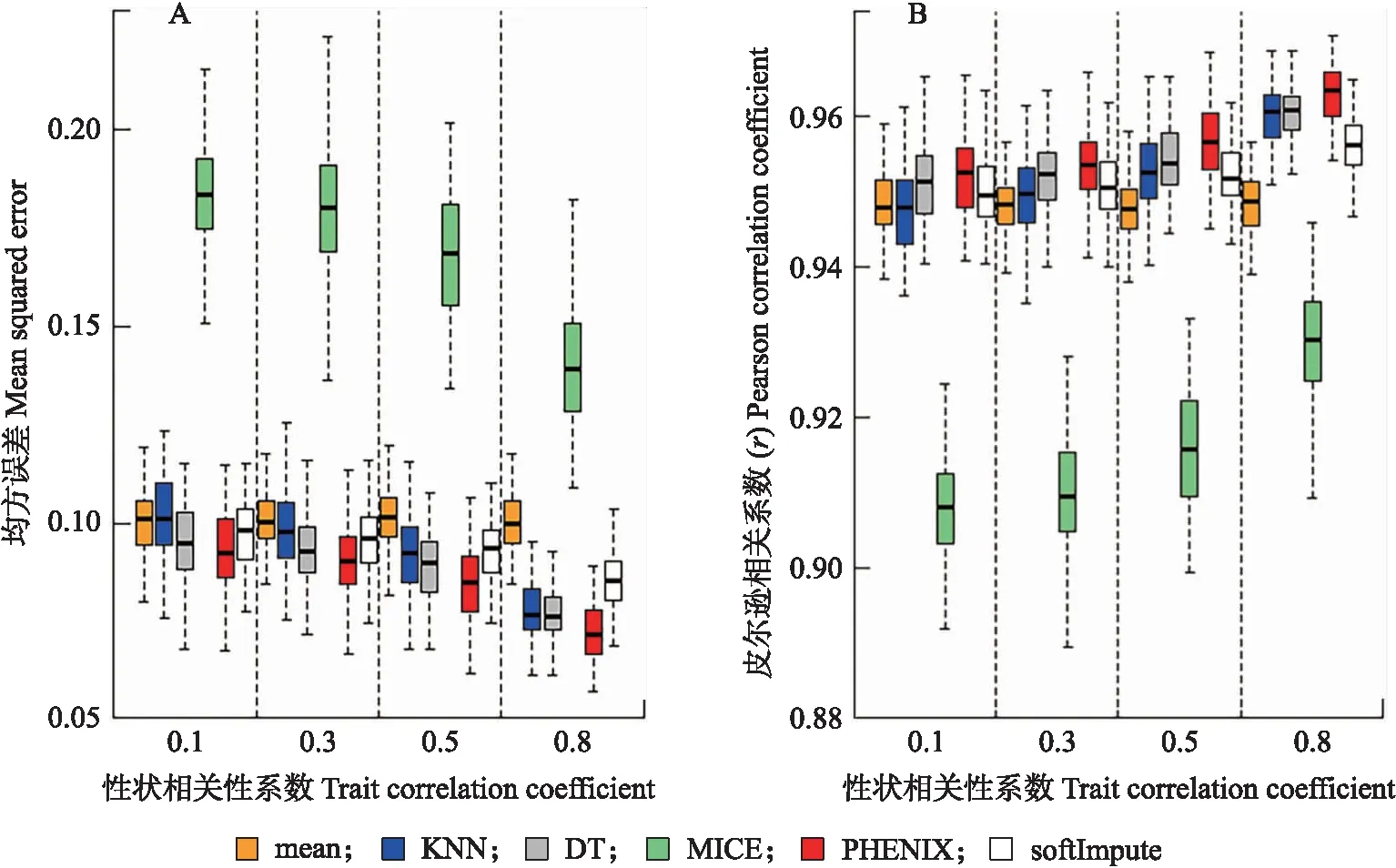
图4 不同性状相关性下mean、KNN、DT、MICE、PHENIX和softImpute插补值与观测值的均方误差(A)和皮尔逊相关系数(B)(重复100次)
从模拟试验的计算时间(表2)可见:mean和softImpute的计算速度最快,均小于2 s,随后是KNN和DT插补法,计算时间为3~40 s,而MICE和PHENIX插补时间相对比较长。由表2也可以看出:数据的表型缺失率和性状相关性对所有方法的运算时间影响均不显著,而随性状数和样本量的增大,运算时间呈现显著增加的趋势。MICE插补法对于性状数较为敏感,随性状数增加,MICE的计算时间呈现出指数级的增长;而PHENIX插补法对于样本量较为敏感,计算时间与样本量呈现出指数级增长的关系。
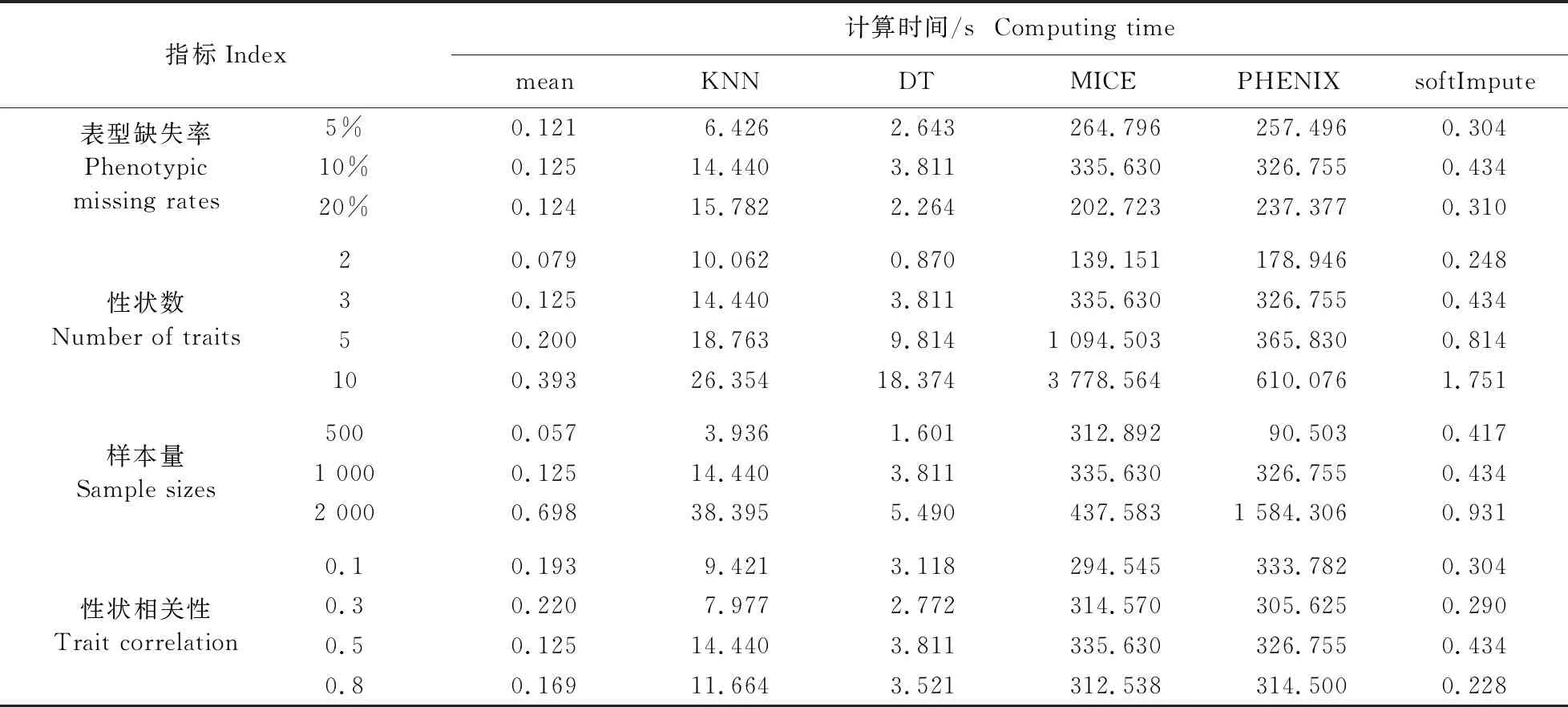
表2 4种模拟研究100次模拟数据集的mean、KNN、DT、MICE、PHENIX和softImpute的计算时间
2.2 真实数据的缺失数据插补
真实拟南芥数据包括199个个体、216 130个SNP,4个与花期相关的性状(LD、SD、LDV和SDV),其表型缺失率分别为16.0%、18.6%、15.6%和20.1%,4个性状之间具有较强的相关性,相关系数为0.65~0.85。
采用mean、KNN、DT、MICE、PHENIX和softImpute插补方法对真实数据进行联合插补,并对插补后的数据进行全基因组关联分析。利用TAIR基因库验证位于显著SNP位点(Bonferroni校正)附近20 kb的关联基因,挖掘出与花期相关的已验证基因。由图5可以看出:PHENIX、DT和KNN检测的已验证基因数最多,分别为22、18和18,并且共同检测到超过17个已验证基因,说明3种方法能够利用拟南芥真实数据的复杂遗传结构,得到更接近真实情况的插补值。mean检测的已验证基因数为14,并且与其他方法共同检测的基因较多,softImpute和MICE检测的已验证基因数较少,分别为7和2。

图5 利用mean、KNN、DT、MICE、PHENIX和softImpute插补数据和观测数据检测以及共同检测的已验证基因数
6种插补方法中,mean、KNN、DT和softImpute插补法的计算时间在同一个数量级上,均小于0.1 s,PHENIX插补法计算时间略长,约0.4 s,而MICE插补法长于其他5种方法,为3.6 s,这与模拟数据的计算时间一致。由分析结果、检测准确性和运算时间等指标可以看出,PHENIX、DT和KNN多性状联合分析插补法可以充分利用性状之间的相关性,插补效果优于其余方法,并适宜在真实数据分析中进行表型缺失的插补。
2.3 数量性状表型插补前后关联分析结果的比较
表型数据插补能够利用现有观测数据信息,预测缺失值以产生完整数据,避免直接删除数据带来的信息损失,增大样本量,提高后续研究的准确性。因此,本研究对拟南芥实际数据中表型数据插补前、后的结果进行比较,利用关联分析检测到的显著SNP进行已验证基因筛选,原始观测数据筛选的已验证基因数为15,少于PHENIX、KNN和DT方法,比mean、softImpute和MICE方法检测到的已验证基因数多(图5)。这进一步说明了PHENIX、DT和KNN插补方法得到的缺失数据估计值更加准确、更接近真实值,因此,其关联分析结果功效更高、检测的已验证基因数更多,而mean、softImpute和MICE方法的结果不能令人满意。
3 结论与讨论
数据缺失是进行全基因组关联分析时普遍存在且难以避免的问题,不但增大了统计分析难度,而且还会造成分析结果的偏倚,使结果无法准确反映总体的真实特征,甚至得出错误的分析结果。对于不同类型的缺失数据,选择合适的预测方法能够为后续分析结果的准确性和可靠性奠定基础。因此,比较不同插补方法处理不同缺失样本的效果具有重要意义。
本研究针对不同研究目的生成4组模拟数据集,采用mean、KNN、DT、MICE、PHENIX和softImpute插补方法,分析多性状的表型缺失率、性状数、样本量和性状相关性对表型插补准确性的影响。模拟研究结果显示,在不同的试验中PHENIX、KNN和DT的插补效果较好,具有较小的MSE和较高的r;其次是softImpute插补方法;而mean插补法仅仅利用了表型的平均值,降低了表型的变异程度,表现不佳;MICE插补法的MSE最大、r最小,其结果不能令人满意。同时,利用6种插补方法对实际缺失数据进行多性状联合插补,并通过全基因组关联分析方法验证了插补方法的可靠性。结果显示多性状联合插补方法PHENIX、KNN和DT可以利用性状之间的遗传结构,检测到的已验证基因数最多,并且它们共同检测到的基因较多。
从计算时间的角度来看,mean、KNN、DT和softImpute插补法在模拟分析和实际数据分析时均比较快,MICE和PHENIX插补法的计算时间相对较长,MICE插补法的计算时间受性状数的影响比较大,而PHENIX插补法受样本量的影响比较大。
不同的缺失数据处理方法有不同的特点,进行插补方法选择时,应尽量全面了解数据的背景和特征,尽可能利用各种辅助信息来进行缺失值的预测插补,如性状之间的相关性等,有利于提高缺失数据的插补准确性。
猜你喜欢
心理学报(2022年10期)2022-10-12
福建农林大学学报(自然科学版)(2022年5期)2022-10-08
中国现代医生(2022年21期)2022-08-22
中国循证心血管医学杂志(2022年1期)2022-03-15
内蒙古统计(2021年4期)2021-12-06
建材发展导向(2021年10期)2021-07-16
新世纪智能(语文备考)(2021年11期)2021-03-08
天津医科大学学报(2021年1期)2021-01-26
医药前沿(2020年20期)2020-11-10
三农资讯半月报(2020年2期)2020-03-09
